

Contributions
Abstract: EP898
Type: E-Poster Presentation
Session title: Myelodysplastic syndromes - Biology & Translational Research
Background
Unclassifiable myelodysplastic syndrome defined by a cytogenetic abnormality (MDS-U-C) is a rare, poorly characterized entity that encompasses patients with at least one cytopenia, no marked dysplasia and a karyotype with one or more abnormalities considered to be presumptive criteria for MDS. Many cytopenia are still referred to as idiopathic cytopenia of undetermined significance (ICUS) because the cytological and cytogenetic diagnostic criteria for MDS are not met. Thanks to next generation sequencing (NGS) techniques, the discovery of somatic mutations in over 30% of patients with ICUS has prompted the definition of the entity “clonal cytopenia of undetermined significance”(CCUS). At present, these somatic abnormalities are not included in the WHO’s diagnostic classification of MDS.
Aims
The aim of this study was to determine the clinical, laboratory and genetic characteristics of patients with cytopenia and cytogenetic abnormalities (regardless of whether or not the latter are listed by the WHO as presumptive) but no significant dysplasia. The ultimate objective was therefore to diagnose MDS-U-C more reliably in real life.
Methods
We performed a retrospective study of 64 patients diagnosed between 2011 and 2019 in nine French centres. The main inclusion criteria were cytopenia, at least one cytogenetic abnormality and the absence of marked dysplasia. Bone marrow smears were examined independently by three cytologists. Karyotypes were reviewed by members of the Groupe Francophone de Cytogénétique Hématologique. NGS was used to screen patients for a targeted panel of 58 genes frequently altered in MDS.
Results
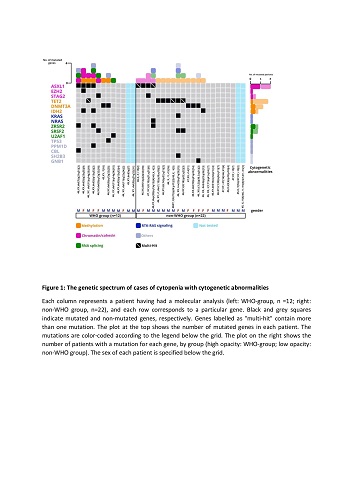
The centralized cytology review led to 30 of the 64 patients being reclassified as having MDS; this relatively high proportion reflects the poor specificity of dysplastic features. We then divided the remaining 34 dysplasia-free patients into two groups: the “WHO group” comprising 12 individuals with a presumptive cytogenetic abnormality (including del(5q) (n=4), del(11q) (n=3), del(9q) (n=3), del(7q) (n=1) and -7 (n=1); median age: 74.3) and the “non-WHO group” comprising 22 individuals with non-recurrent abnormalities (except for trisomy 15 (n=5); median age: 76.7). The proportion of patients with one or more somatic mutations was 80% in the WHO group (mean number per patient: 2.4) and 70% in the non-WHO group (mean: 1.4) (p=0.11). The frequency of ASXL1 mutations was higher in the WHO group (40% vs. 20 % in the non-WHO group; p=0.38), whereas the frequency of TET2 mutations was higher in the non-WHO group (30% vs 10% in the WHO group; p=0.37) (Figure 1). The number and type of mutations in the WHO group were similar to those seen in patients with MDS, who harboured a median of 2 or 3 driver mutations (predominantly in TET2, DNMT3A and ASXL1). Although the same genes were involved in the non-WHO group, the mutation burden was lower than in MDS but higher than in clonal haematopoiesis of indeterminate potential (CHIP). With a median follow-up time of 10.4 months, the WHO group appeared to have a worse prognosis: 25% of these patients progressed to high-grade MDS (vs.4.5% in the non-WHO group; p=0.11).
Conclusion
Our present results confirmed that MDS-U-C is rare and is difficult to diagnose. Even though patients meeting all the criteria for MDS-U-C (i.e. the WHO group) seemed to progress more often to high-risk MDS and harboured a greater number of deleterious mutations (e.g. mutations in ASXL1), “non-WHO” patients must not be neglected; the latter require close monitoring and should perhaps be classified as cases of CCUS.
Keyword(s): Cytogenetic abnormalities, Myelodysplasia
Abstract: EP898
Type: E-Poster Presentation
Session title: Myelodysplastic syndromes - Biology & Translational Research
Background
Unclassifiable myelodysplastic syndrome defined by a cytogenetic abnormality (MDS-U-C) is a rare, poorly characterized entity that encompasses patients with at least one cytopenia, no marked dysplasia and a karyotype with one or more abnormalities considered to be presumptive criteria for MDS. Many cytopenia are still referred to as idiopathic cytopenia of undetermined significance (ICUS) because the cytological and cytogenetic diagnostic criteria for MDS are not met. Thanks to next generation sequencing (NGS) techniques, the discovery of somatic mutations in over 30% of patients with ICUS has prompted the definition of the entity “clonal cytopenia of undetermined significance”(CCUS). At present, these somatic abnormalities are not included in the WHO’s diagnostic classification of MDS.
Aims
The aim of this study was to determine the clinical, laboratory and genetic characteristics of patients with cytopenia and cytogenetic abnormalities (regardless of whether or not the latter are listed by the WHO as presumptive) but no significant dysplasia. The ultimate objective was therefore to diagnose MDS-U-C more reliably in real life.
Methods
We performed a retrospective study of 64 patients diagnosed between 2011 and 2019 in nine French centres. The main inclusion criteria were cytopenia, at least one cytogenetic abnormality and the absence of marked dysplasia. Bone marrow smears were examined independently by three cytologists. Karyotypes were reviewed by members of the Groupe Francophone de Cytogénétique Hématologique. NGS was used to screen patients for a targeted panel of 58 genes frequently altered in MDS.
Results
The centralized cytology review led to 30 of the 64 patients being reclassified as having MDS; this relatively high proportion reflects the poor specificity of dysplastic features. We then divided the remaining 34 dysplasia-free patients into two groups: the “WHO group” comprising 12 individuals with a presumptive cytogenetic abnormality (including del(5q) (n=4), del(11q) (n=3), del(9q) (n=3), del(7q) (n=1) and -7 (n=1); median age: 74.3) and the “non-WHO group” comprising 22 individuals with non-recurrent abnormalities (except for trisomy 15 (n=5); median age: 76.7). The proportion of patients with one or more somatic mutations was 80% in the WHO group (mean number per patient: 2.4) and 70% in the non-WHO group (mean: 1.4) (p=0.11). The frequency of ASXL1 mutations was higher in the WHO group (40% vs. 20 % in the non-WHO group; p=0.38), whereas the frequency of TET2 mutations was higher in the non-WHO group (30% vs 10% in the WHO group; p=0.37) (Figure 1). The number and type of mutations in the WHO group were similar to those seen in patients with MDS, who harboured a median of 2 or 3 driver mutations (predominantly in TET2, DNMT3A and ASXL1). Although the same genes were involved in the non-WHO group, the mutation burden was lower than in MDS but higher than in clonal haematopoiesis of indeterminate potential (CHIP). With a median follow-up time of 10.4 months, the WHO group appeared to have a worse prognosis: 25% of these patients progressed to high-grade MDS (vs.4.5% in the non-WHO group; p=0.11).
Conclusion
Our present results confirmed that MDS-U-C is rare and is difficult to diagnose. Even though patients meeting all the criteria for MDS-U-C (i.e. the WHO group) seemed to progress more often to high-risk MDS and harboured a greater number of deleterious mutations (e.g. mutations in ASXL1), “non-WHO” patients must not be neglected; the latter require close monitoring and should perhaps be classified as cases of CCUS.
Keyword(s): Cytogenetic abnormalities, Myelodysplasia