

Contributions
Abstract: EP875
Type: E-Poster Presentation
Session title: Lymphoma Biology & Translational Research
Background
Adult T-cell leukemia/ lymphoma (ATL) is derived from mature T-cells transformed by human T-cell lymphotropic virus type 1 (HTLV-1). Following HTLV-1 infection, it takes decades for carriers to develop ATL. HTLV-1–infected T-cells undergo multi-step leukemogenesis, with accumulation of genetic and epigenetic abnormalities. Trimethylation at histone H3Lys27 (H3K27me3) catalyzed by polycomb-repressive complex 2 accumulates aberrantly in HTLV-1-infected T-cells and its accumulation correlates with ATL development and progression.
We recently identified common differentially hypermethylated positions that are specific to HTLV-1-infected T-cells using comprehensive DNA methylation analysis. The accumulation of DNA methylation at these sites correlated strongly with ATL development and progression (Blood 2020, 136: 871-884). OR-2100, which we developed as a novel decitabine prodrug with enhanced oral bioavailability, suppressed tumor cell growth and global DNA hypomethylation. Taken together, epigenetic abnormalities, especially hypermethylation of DNA and histone H3Lys27, are functionally important for ATL leukemogenesis and represent effective therapeutic targets.
Aims
Here, we aimed to understand how these epigenetic abnormalities accumulate in HTLV-1-infected cells during multi-step carcinogenesis.
Methods
HTLV-1-infected cell lines were cultured in RPMI-1640 medium containing 100 μM methionine. We also prepared modified RPMI-1640 medium with a reduced concentration of methionine. DNA methylation at long interspersed nuclear element-1 (LINE-1), which is a surrogate marker for global DNA methylation status, was determined by bisulfite pyrosequencing. N6-methyladenosine (m6A) in mRNA (m6A RNA methylation) was quantified by ELISA.
Results
Recently, the epigenetic reprogramming that drives T cell differentiation and proliferation was shown to be regulated by the incorporation of extracellular methionine through the amino acid transporter SLC7A5, the expression of which is induced by activation of the T-cell receptor (TCR) signaling pathway. Since several negative regulators of the TCR pathway are silenced by promoter hypermethylation in HTLV-1-infected cells, we hypothesized that epigenetic abnormalities are maintained through sustained SLC7A5-mediated incorporation of methionine in ATL cells.
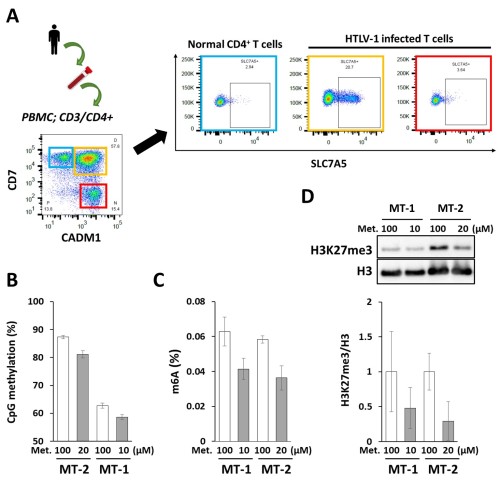
As expected, we found that SLC7A5 gene expression was higher in HTLV-1-infected T-cells than in normal T-cells using the gene expression datasets GSE55851 and GSE33615. Flow cytometric analysis of SLC7A5 protein expression in peripheral blood mononuclear cells from ATL patients revealed SLC7A5 expression in HTLV-1-infected cells from one of 10 ATL patients (Fig. A). Two HTLV-1-infected cell lines, MT-2 and MT-4, also expressed high levels of SLC7A5 protein. Methionine restriction suppressed the growth of HTLV-1-infected cell lines. As expected, DNA and RNA hypomethylation was induced by methionine restriction (Fig. B, C). A decrease in H3K27me3 and protein arginine methylation was also confirmed by western blot analysis (Fig. D). To understand better the epigenetic responses to methionine restriction, we are performing multi-omics analysis using transcriptome, DNA methylome, histone methylome and metabolome data.
Conclusion
Based on these results, we think that the epigenetic state of some HTLV-1-infected cells is regulated by methionine metabolism. Targeting methionine incorporation or metabolism to change epigenetic reprogramming could constitute a novel therapeutic strategy for the treatment of ATL.
Keyword(s): ATL, Epigenetic, Leukemogenesis, Methylation
Abstract: EP875
Type: E-Poster Presentation
Session title: Lymphoma Biology & Translational Research
Background
Adult T-cell leukemia/ lymphoma (ATL) is derived from mature T-cells transformed by human T-cell lymphotropic virus type 1 (HTLV-1). Following HTLV-1 infection, it takes decades for carriers to develop ATL. HTLV-1–infected T-cells undergo multi-step leukemogenesis, with accumulation of genetic and epigenetic abnormalities. Trimethylation at histone H3Lys27 (H3K27me3) catalyzed by polycomb-repressive complex 2 accumulates aberrantly in HTLV-1-infected T-cells and its accumulation correlates with ATL development and progression.
We recently identified common differentially hypermethylated positions that are specific to HTLV-1-infected T-cells using comprehensive DNA methylation analysis. The accumulation of DNA methylation at these sites correlated strongly with ATL development and progression (Blood 2020, 136: 871-884). OR-2100, which we developed as a novel decitabine prodrug with enhanced oral bioavailability, suppressed tumor cell growth and global DNA hypomethylation. Taken together, epigenetic abnormalities, especially hypermethylation of DNA and histone H3Lys27, are functionally important for ATL leukemogenesis and represent effective therapeutic targets.
Aims
Here, we aimed to understand how these epigenetic abnormalities accumulate in HTLV-1-infected cells during multi-step carcinogenesis.
Methods
HTLV-1-infected cell lines were cultured in RPMI-1640 medium containing 100 μM methionine. We also prepared modified RPMI-1640 medium with a reduced concentration of methionine. DNA methylation at long interspersed nuclear element-1 (LINE-1), which is a surrogate marker for global DNA methylation status, was determined by bisulfite pyrosequencing. N6-methyladenosine (m6A) in mRNA (m6A RNA methylation) was quantified by ELISA.
Results
Recently, the epigenetic reprogramming that drives T cell differentiation and proliferation was shown to be regulated by the incorporation of extracellular methionine through the amino acid transporter SLC7A5, the expression of which is induced by activation of the T-cell receptor (TCR) signaling pathway. Since several negative regulators of the TCR pathway are silenced by promoter hypermethylation in HTLV-1-infected cells, we hypothesized that epigenetic abnormalities are maintained through sustained SLC7A5-mediated incorporation of methionine in ATL cells.
As expected, we found that SLC7A5 gene expression was higher in HTLV-1-infected T-cells than in normal T-cells using the gene expression datasets GSE55851 and GSE33615. Flow cytometric analysis of SLC7A5 protein expression in peripheral blood mononuclear cells from ATL patients revealed SLC7A5 expression in HTLV-1-infected cells from one of 10 ATL patients (Fig. A). Two HTLV-1-infected cell lines, MT-2 and MT-4, also expressed high levels of SLC7A5 protein. Methionine restriction suppressed the growth of HTLV-1-infected cell lines. As expected, DNA and RNA hypomethylation was induced by methionine restriction (Fig. B, C). A decrease in H3K27me3 and protein arginine methylation was also confirmed by western blot analysis (Fig. D). To understand better the epigenetic responses to methionine restriction, we are performing multi-omics analysis using transcriptome, DNA methylome, histone methylome and metabolome data.
Conclusion
Based on these results, we think that the epigenetic state of some HTLV-1-infected cells is regulated by methionine metabolism. Targeting methionine incorporation or metabolism to change epigenetic reprogramming could constitute a novel therapeutic strategy for the treatment of ATL.
Keyword(s): ATL, Epigenetic, Leukemogenesis, Methylation