

Contributions
Abstract: EP708
Type: E-Poster Presentation
Session title: Enzymopathies, membranopathies and other anemias
Background
Hereditary haemolytic anemias (HA) are a group of heterogeneous, low frequency entities characterized by the premature destruction of red blood cells (RBC). They are usually secondary to genetic mutations leading to alterations in hemoglobin or membrane structure and enzimatic production, as well as RBC differenciation and proliferation. In many cases, attaining an accurate diagnosis is complex due to overlapping clinical presentation, variable clinical expression and great variety of implicated genes.
The recent use of next generation sequencing (NGS) has led to an improvement in our understanding of these entities by widening the knowledge of genetic alterations involved, and has proved a useful tool for both diagnosis and genetic advice.
Aims
The aim of this study is to evaluate the results of an NGS panel for congenital anemias and medullary aplasia in a single center in Madrid, Spain.
Methods
A total of 39 patients were included in the study, 71% had clinical suspicion of HA and 28% had familiar history of hereditary anemia. We performed NGS (Ion Torrent S5XL System – Thermofisher Scientific) of a panel comprising 55 genes related to metabolic enzymes (AK1, ALDOA, BPGM, G6PD, GPI, GSS, HK1, NT5C3A, PFKFB1, PGK, PKLR, TPI1), membrane and cytoskeleton proteins (ANK1, EPB41, EPB42, EPB72, SLC4A1, SPTA1, SPTB, PIEZO1, PIGA, PIGT), ribosomic proteins (RPL11, RPL15, RPL5, RPS19, RPS26, SBDS), telomeric regulators (RTEL1,TERT, TERC, TINF2, DKC1), DNA repair (FANCA, FANCC, FANCG, LIG4), transcription factors (GATA1, GATA2, MECOM), globin chains (HBA1, HBA2, HBB) and other non-classifiable genes (ANKRD26, ATRX1, DNAJC21, SRP72, TRS2, HAX1, SAMD9, SAMDL9, ADAMTS13, ELANE, ERCC6L2, DDX41).
Results
The median age was 31 years; R(0-72), 59% were female. Mean hemoglobin was 12.5 g/dL and mean reticulocyte count was 100650/ml. Hemolysis paramethers were slightly elevated (mean LDH 244 U/L, mean bilirubin 1.73 mg/dL).
81% of the patients presented at least one genetic variant in the NGS study (median of 3 variants per patient). Among these, 71% presented pathogenic/likely pathogenic variants which were considered useful for the diagnosis, and 6 had variants of undetermined significance. Single Nucleotide Variation (SNV) were the most common, present in all patients with any variant. Insertions or deletions (indels) were additionally present in 20% of them.
Most of the variants were detected in genes associated with membrane and cytoskeleton (24; 58.5%), others non-classifiable (10; 24.4%), DNA repair (8; 19.5%), enzymatic genes (6; 14.6%), globin chains (5; 12.2%), telomeric genes and transcription factors (both 4; 9.8%) and ribosomic genes (3; 7.3%).
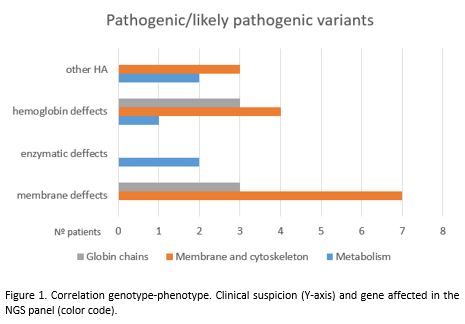
Genotype-phenotype correlation was variable in each group. Erytrocite membrane defects were confirmed in 70% of the patients with this clinical suspicion, but only 38% of the patients who were suspected of an alteration in globin chains had a proven genetic variant in these genes with a high percentage presenting alterations in membrane and cytoskeleton genes. Among the 5 patients with unknown HA, 3 had likely pathogenic variants in membrane and cytoskeleton genes and 2 in metabolic enzymes genes. 5 patients had pathogenic/likely pathogenic variants in various genes pertaining to more than one group, a situation with uncertain clinical significance.
Conclusion
NGS is a useful tool for the diagnosis of hereditary hemolytic anemias. However, there are still a lot of unknowns in the genetic landscape that must be addressed to improve our understanding of these diseases.
Keyword(s): Anemia, Erythrocyte, Inherited disease, Molecular
Abstract: EP708
Type: E-Poster Presentation
Session title: Enzymopathies, membranopathies and other anemias
Background
Hereditary haemolytic anemias (HA) are a group of heterogeneous, low frequency entities characterized by the premature destruction of red blood cells (RBC). They are usually secondary to genetic mutations leading to alterations in hemoglobin or membrane structure and enzimatic production, as well as RBC differenciation and proliferation. In many cases, attaining an accurate diagnosis is complex due to overlapping clinical presentation, variable clinical expression and great variety of implicated genes.
The recent use of next generation sequencing (NGS) has led to an improvement in our understanding of these entities by widening the knowledge of genetic alterations involved, and has proved a useful tool for both diagnosis and genetic advice.
Aims
The aim of this study is to evaluate the results of an NGS panel for congenital anemias and medullary aplasia in a single center in Madrid, Spain.
Methods
A total of 39 patients were included in the study, 71% had clinical suspicion of HA and 28% had familiar history of hereditary anemia. We performed NGS (Ion Torrent S5XL System – Thermofisher Scientific) of a panel comprising 55 genes related to metabolic enzymes (AK1, ALDOA, BPGM, G6PD, GPI, GSS, HK1, NT5C3A, PFKFB1, PGK, PKLR, TPI1), membrane and cytoskeleton proteins (ANK1, EPB41, EPB42, EPB72, SLC4A1, SPTA1, SPTB, PIEZO1, PIGA, PIGT), ribosomic proteins (RPL11, RPL15, RPL5, RPS19, RPS26, SBDS), telomeric regulators (RTEL1,TERT, TERC, TINF2, DKC1), DNA repair (FANCA, FANCC, FANCG, LIG4), transcription factors (GATA1, GATA2, MECOM), globin chains (HBA1, HBA2, HBB) and other non-classifiable genes (ANKRD26, ATRX1, DNAJC21, SRP72, TRS2, HAX1, SAMD9, SAMDL9, ADAMTS13, ELANE, ERCC6L2, DDX41).
Results
The median age was 31 years; R(0-72), 59% were female. Mean hemoglobin was 12.5 g/dL and mean reticulocyte count was 100650/ml. Hemolysis paramethers were slightly elevated (mean LDH 244 U/L, mean bilirubin 1.73 mg/dL).
81% of the patients presented at least one genetic variant in the NGS study (median of 3 variants per patient). Among these, 71% presented pathogenic/likely pathogenic variants which were considered useful for the diagnosis, and 6 had variants of undetermined significance. Single Nucleotide Variation (SNV) were the most common, present in all patients with any variant. Insertions or deletions (indels) were additionally present in 20% of them.
Most of the variants were detected in genes associated with membrane and cytoskeleton (24; 58.5%), others non-classifiable (10; 24.4%), DNA repair (8; 19.5%), enzymatic genes (6; 14.6%), globin chains (5; 12.2%), telomeric genes and transcription factors (both 4; 9.8%) and ribosomic genes (3; 7.3%).
Genotype-phenotype correlation was variable in each group. Erytrocite membrane defects were confirmed in 70% of the patients with this clinical suspicion, but only 38% of the patients who were suspected of an alteration in globin chains had a proven genetic variant in these genes with a high percentage presenting alterations in membrane and cytoskeleton genes. Among the 5 patients with unknown HA, 3 had likely pathogenic variants in membrane and cytoskeleton genes and 2 in metabolic enzymes genes. 5 patients had pathogenic/likely pathogenic variants in various genes pertaining to more than one group, a situation with uncertain clinical significance.
Conclusion
NGS is a useful tool for the diagnosis of hereditary hemolytic anemias. However, there are still a lot of unknowns in the genetic landscape that must be addressed to improve our understanding of these diseases.
Keyword(s): Anemia, Erythrocyte, Inherited disease, Molecular