

Contributions
Abstract: EP616
Type: E-Poster Presentation
Session title: Chronic lymphocytic leukemia and related disorders - Biology & Translational Research
Background
Complex karyotype (CK) is a predictor of poor prognosis in patients with chronic lymphocytic leukemia (CLL). CK is defined as the presence of ≥3 chromosomal aberrations in the same clone by chromosome banding analysis (CBA). However, recent studies have suggested that the best cut-off to predict an adverse outcome in CLL should be established at 5 abnormalities detected by either CBA or chromosomal microarrays (CMA) (Baliakas et al, 2019; Leeksma et al, 2021). Both methods are useful for risk stratification of CLL patients, but they are not equivalent due to limitations intrinsic to each technique (Ramos-Campoy et al, 2021). Optical genome mapping (OGM) is a novel method that relies on the imaging of long DNA molecules (>250 kb) labeled at specific sites. The unique pattern of labels throughout the genome allows to map the genomic location of every molecule and detect both balanced and unbalanced abnormalities with high resolution and sensitivity. This is as promising technology that overcomes most of the limitations of CBA and CMA.
Aims
1. To compare the results obtained by OGM with previous CBA and CMA information. 2. To analyze the usefulness of OGM in the assessment of genomic complexity in CLL patients.
Methods
A total of 22 CLL patients were included, 12 carried CK by CBA and 10 were non-CK. Tumor DNA extracted from peripheral blood was analyzed by OGM (Bionano Genomics). Abnormalities found by OGM were compared with available CBA, FISH and CMA (12 CytoScan HD and 10 CytoScan 750K, ThermoFisher) results, and complexity detected by OGM in each group was assessed.
Results
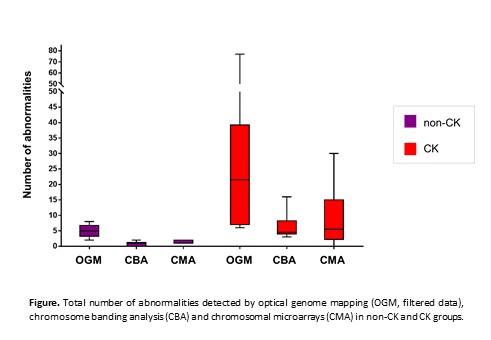
OGM detection rate of previously known abnormalities was 93% (14/15) and 86% (119/138) in non-CK and CK groups, respectively. Discordances were mainly small copy number alterations (CNAs) detected by CMA and subclonal aberrations in the karyotype expanded during CBA culture but represented in a minor proportion, also below CMA sensitivity. Size and coordinates of CNAs detected by OGM were highly concordant with those defined by CMA. Moreover, OGM provided additional structural information associated with some microarray CNAs or helped to identify unknown cytogenetic elements reported by CBA in 11/22 (50%) patients. Additional new abnormalities, including balanced rearrangements and CNAs, were found by OGM in both groups. The median number of abnormalities by OGM was 72 (range: 38-103) in non-CK and 120 (range: 50-249) in CK patients. They were highly enriched in insertions, duplications or deletions of unknown clinical significance displaying sizes smaller than CMA resolution (from only 3 to 100Kbp). When polymorphic variants and unbalanced abnormalities smaller than 100Kbp were filtered, CK group displayed significantly a more complex genome than non-CK patients [median 22 (6-77) vs 5 (2-8), P<0.01]. Globally, OGM detected a higher complexity, although a good correlation between the number of aberrations recorded by OGM and CBA or CMA was observed (rp=0.831 and rp=0.935, respectively) (Figure).
Conclusion
1. OGM is a valuable tool to assess genomic complexity in CLL. It effectively detects the vast majority of the abnormalities defined by standard methods, allowing a comprehensive interpretation of some of the alterations; 2. Several additional abnormalities of unknown clinical significance are identified by OGM; 3. Further studies are required to define genomic complexity criteria by OGM and implement this technique in the routine management of CLL patients.
Keyword(s): Chronic lymphocytic leukemia, Complex aberrant karyotype, Cytogenetic abnormalities
Abstract: EP616
Type: E-Poster Presentation
Session title: Chronic lymphocytic leukemia and related disorders - Biology & Translational Research
Background
Complex karyotype (CK) is a predictor of poor prognosis in patients with chronic lymphocytic leukemia (CLL). CK is defined as the presence of ≥3 chromosomal aberrations in the same clone by chromosome banding analysis (CBA). However, recent studies have suggested that the best cut-off to predict an adverse outcome in CLL should be established at 5 abnormalities detected by either CBA or chromosomal microarrays (CMA) (Baliakas et al, 2019; Leeksma et al, 2021). Both methods are useful for risk stratification of CLL patients, but they are not equivalent due to limitations intrinsic to each technique (Ramos-Campoy et al, 2021). Optical genome mapping (OGM) is a novel method that relies on the imaging of long DNA molecules (>250 kb) labeled at specific sites. The unique pattern of labels throughout the genome allows to map the genomic location of every molecule and detect both balanced and unbalanced abnormalities with high resolution and sensitivity. This is as promising technology that overcomes most of the limitations of CBA and CMA.
Aims
1. To compare the results obtained by OGM with previous CBA and CMA information. 2. To analyze the usefulness of OGM in the assessment of genomic complexity in CLL patients.
Methods
A total of 22 CLL patients were included, 12 carried CK by CBA and 10 were non-CK. Tumor DNA extracted from peripheral blood was analyzed by OGM (Bionano Genomics). Abnormalities found by OGM were compared with available CBA, FISH and CMA (12 CytoScan HD and 10 CytoScan 750K, ThermoFisher) results, and complexity detected by OGM in each group was assessed.
Results
OGM detection rate of previously known abnormalities was 93% (14/15) and 86% (119/138) in non-CK and CK groups, respectively. Discordances were mainly small copy number alterations (CNAs) detected by CMA and subclonal aberrations in the karyotype expanded during CBA culture but represented in a minor proportion, also below CMA sensitivity. Size and coordinates of CNAs detected by OGM were highly concordant with those defined by CMA. Moreover, OGM provided additional structural information associated with some microarray CNAs or helped to identify unknown cytogenetic elements reported by CBA in 11/22 (50%) patients. Additional new abnormalities, including balanced rearrangements and CNAs, were found by OGM in both groups. The median number of abnormalities by OGM was 72 (range: 38-103) in non-CK and 120 (range: 50-249) in CK patients. They were highly enriched in insertions, duplications or deletions of unknown clinical significance displaying sizes smaller than CMA resolution (from only 3 to 100Kbp). When polymorphic variants and unbalanced abnormalities smaller than 100Kbp were filtered, CK group displayed significantly a more complex genome than non-CK patients [median 22 (6-77) vs 5 (2-8), P<0.01]. Globally, OGM detected a higher complexity, although a good correlation between the number of aberrations recorded by OGM and CBA or CMA was observed (rp=0.831 and rp=0.935, respectively) (Figure).
Conclusion
1. OGM is a valuable tool to assess genomic complexity in CLL. It effectively detects the vast majority of the abnormalities defined by standard methods, allowing a comprehensive interpretation of some of the alterations; 2. Several additional abnormalities of unknown clinical significance are identified by OGM; 3. Further studies are required to define genomic complexity criteria by OGM and implement this technique in the routine management of CLL patients.
Keyword(s): Chronic lymphocytic leukemia, Complex aberrant karyotype, Cytogenetic abnormalities