

Contributions
Abstract: EP610
Type: E-Poster Presentation
Session title: Chronic lymphocytic leukemia and related disorders - Biology & Translational Research
Background
Genomic and transcriptomic studies in chronic lymphocytic leukaemia (CLL) have significantly improved our understanding of disease biology and drug resistance mechanisms whilst simultaneously identifying new biomarkers. Despite the ability of such biomarkers to predict treatment response and survival, the prognostic/predictive information they provide is not absolute. Since disease phenotype is determined at the protein level and given the imperfect correlation between mRNA and protein expression, we hypothesised that a proteomic approach might shed new light on pathogenetic mechanisms and indicate potential drug targets and biomarkers.
Aims
Using SWATH-MS (Sequential Window Acquisition of All Theoretical Mass Spectra), the study aimed to elucidate differences in the pre-treatment CLL proteome that correlated with response to fludarabine-based chemo-immunotherapy defined by post-treatment minimal residual disease (MRD) status.
Methods
Pre-treatment cryopreserved peripheral blood mononuclear cells collected from patients as part of the ARCTIC and AdMIRe trials were lysed and protein extracted using published protocols. After trypsin digestion, the peptides were processed and mass spectrometry (Sciex TripleTOF 6600) performed. Data was normalized within DIA-NN software and proteins were identified (q<0.01) using our published CLL-specific SWATH library. Data was visualized in R and the inter-batch variation removed along with an outlier. Differential protein expression analysis was performed between MRD positive (MRD+, defined as >1 CLL cell in 10,000 leukocytes in the bone marrow) and negative (MRD-) groups measured three months after the end of treatment. Receiver operating characteristic (ROC) analysis was performed to split patients into low- and high-expression groups for each protein using thresholds that gave the optimum sensitivity and specificity. Functional analyses were performed using Ingenuity Pathway Analysis and Gene Set Enrichment Analysis tools.
Results
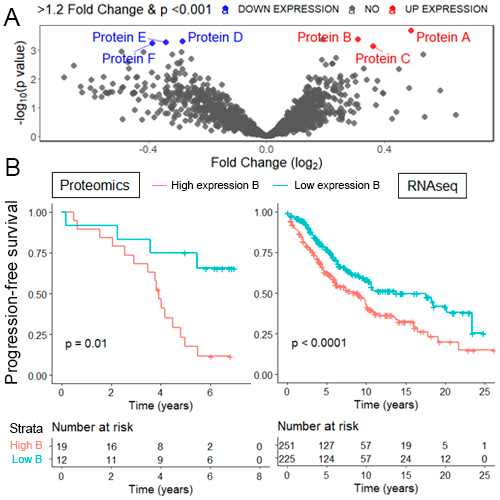
Thirty-two patients were included in the study. Bone marrow MRD data was available for 29 patients, of which 17 (59%) were MRD+ and 12 (41%) MRD-. The median age was 64 (IQR 59-68.5), with a male preponderance (75%). Of the 4696 proteins identified, 2357 were consistently detected and used for subsequent analysis. Using >1.2 fold change and p<0.001 cut-offs, six differentially expressed proteins were identified (proteins A-F; Fig. A). Unsupervised clustering and principal component analysis plots of the six proteins showed clustering of samples based on their MRD status. In univariate analysis, high expression of protein B along with a low expression of proteins D and E were associated with increased risk of progression or death (HR 4.17 [p=0.012], HR 2.75 [p=0.041] and HR 2.53 [p=0.046], respectively). The findings were corroborated using an external RNAseq dataset of 476 patients (The International Cancer Genome Consortium), which confirmed that protein B and its mRNA expression were associated with worse progression-free survival (PFS) in both datasets (p=0.01 and p<0.0001; Fig. B). Functional analyses of proteomic data revealed enrichment of pathways related to mTOR along with processes related to Golgi vesicle transport and protein localization to endoplasmic reticulum, suggesting a possible drug resistance mechanism.
Conclusion
Proteomic analysis using SWATH-MS provides a novel approach to elucidating pathogenetic mechanisms in CLL. In this study, protein B was identified as a potential drug target and biomarker for therapy response.
Keyword(s): Chronic lymphocytic leukemia, Prognostic, Proteomics, Resistance
Abstract: EP610
Type: E-Poster Presentation
Session title: Chronic lymphocytic leukemia and related disorders - Biology & Translational Research
Background
Genomic and transcriptomic studies in chronic lymphocytic leukaemia (CLL) have significantly improved our understanding of disease biology and drug resistance mechanisms whilst simultaneously identifying new biomarkers. Despite the ability of such biomarkers to predict treatment response and survival, the prognostic/predictive information they provide is not absolute. Since disease phenotype is determined at the protein level and given the imperfect correlation between mRNA and protein expression, we hypothesised that a proteomic approach might shed new light on pathogenetic mechanisms and indicate potential drug targets and biomarkers.
Aims
Using SWATH-MS (Sequential Window Acquisition of All Theoretical Mass Spectra), the study aimed to elucidate differences in the pre-treatment CLL proteome that correlated with response to fludarabine-based chemo-immunotherapy defined by post-treatment minimal residual disease (MRD) status.
Methods
Pre-treatment cryopreserved peripheral blood mononuclear cells collected from patients as part of the ARCTIC and AdMIRe trials were lysed and protein extracted using published protocols. After trypsin digestion, the peptides were processed and mass spectrometry (Sciex TripleTOF 6600) performed. Data was normalized within DIA-NN software and proteins were identified (q<0.01) using our published CLL-specific SWATH library. Data was visualized in R and the inter-batch variation removed along with an outlier. Differential protein expression analysis was performed between MRD positive (MRD+, defined as >1 CLL cell in 10,000 leukocytes in the bone marrow) and negative (MRD-) groups measured three months after the end of treatment. Receiver operating characteristic (ROC) analysis was performed to split patients into low- and high-expression groups for each protein using thresholds that gave the optimum sensitivity and specificity. Functional analyses were performed using Ingenuity Pathway Analysis and Gene Set Enrichment Analysis tools.
Results
Thirty-two patients were included in the study. Bone marrow MRD data was available for 29 patients, of which 17 (59%) were MRD+ and 12 (41%) MRD-. The median age was 64 (IQR 59-68.5), with a male preponderance (75%). Of the 4696 proteins identified, 2357 were consistently detected and used for subsequent analysis. Using >1.2 fold change and p<0.001 cut-offs, six differentially expressed proteins were identified (proteins A-F; Fig. A). Unsupervised clustering and principal component analysis plots of the six proteins showed clustering of samples based on their MRD status. In univariate analysis, high expression of protein B along with a low expression of proteins D and E were associated with increased risk of progression or death (HR 4.17 [p=0.012], HR 2.75 [p=0.041] and HR 2.53 [p=0.046], respectively). The findings were corroborated using an external RNAseq dataset of 476 patients (The International Cancer Genome Consortium), which confirmed that protein B and its mRNA expression were associated with worse progression-free survival (PFS) in both datasets (p=0.01 and p<0.0001; Fig. B). Functional analyses of proteomic data revealed enrichment of pathways related to mTOR along with processes related to Golgi vesicle transport and protein localization to endoplasmic reticulum, suggesting a possible drug resistance mechanism.
Conclusion
Proteomic analysis using SWATH-MS provides a novel approach to elucidating pathogenetic mechanisms in CLL. In this study, protein B was identified as a potential drug target and biomarker for therapy response.
Keyword(s): Chronic lymphocytic leukemia, Prognostic, Proteomics, Resistance