

Contributions
Abstract: EP578
Type: E-Poster Presentation
Session title: Bleeding disorders (congenital and acquired)
Background
Mild hemophilia A (HA) and B (HB) are X-linked recessive hereditary diseases, characterized by factor VIII and IX deficiency, respectively. Patients with mild or moderate hemophilia mostly present specific mutations in F8 gene (Xq28) or F9 (Xq22), detectable by direct sequencing. More than 4000 mutations have been detected so far and genotype analysis should be offered in selected patients. Therefore, genetic assessment is important to provide reproductive options.
Aims
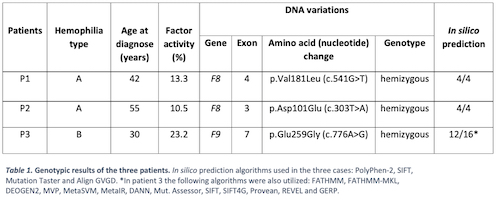
We report three new likely pathogenic variants in three male Spanish patients. Patient 1 presented asymptomatic prolonged pre-operatory aPTT and the others were studied due to hemorrhagic complications of surgery. All presented isolated reduced factor VIII or IX activity levels.
Methods
Molecular genetic studies: DNA was extracted from peripheral blood. Coding exons and adjacent intronic regions of F8 and F9 gene were amplified by polymerase chain reaction (PCR) and sequenced on Applied Biosystems® 3500DX Genetic Analyzer. The results were checked in HGMD, ClinVar, dbSNP, gnomAD, and 1000G international databases and different in silico prediction algorithms, such as: PolyPhen-2, SIFT, Mutation Taster, Align GVGD, REVEL and others were consulted.
Results
Three new missense variants were found (table 1). These variants have not been previously reported in the literature or in any of the databases consulted. They do affect the physical-chemical characteristics of the protein (in silico prediction). Therefore, they are classified as likely pathogenic. Because of this, we extended the study to first-degree relatives; finding carriers that were not diagnose at that time. A 20-year-old daughter (patient 1) and a sister (patient 2) received genetic counseling.
Conclusion
These new genetic variants confirm the clinical suspicion of mild hemophilia. The molecular genetic analysis in the family members helps finding asymptomatic carriers so we can provide them genetic counseling. Future reproductive options will be considered in the youngest patient and any family members if needed, firstly and foremostly, prenatal fetal sex chromosome testing by maternal plasma DNA sequencing on female carriers.
Keyword(s): Diagnosis, Gene expression, Hemophilia A, Hemophilia B
Abstract: EP578
Type: E-Poster Presentation
Session title: Bleeding disorders (congenital and acquired)
Background
Mild hemophilia A (HA) and B (HB) are X-linked recessive hereditary diseases, characterized by factor VIII and IX deficiency, respectively. Patients with mild or moderate hemophilia mostly present specific mutations in F8 gene (Xq28) or F9 (Xq22), detectable by direct sequencing. More than 4000 mutations have been detected so far and genotype analysis should be offered in selected patients. Therefore, genetic assessment is important to provide reproductive options.
Aims
We report three new likely pathogenic variants in three male Spanish patients. Patient 1 presented asymptomatic prolonged pre-operatory aPTT and the others were studied due to hemorrhagic complications of surgery. All presented isolated reduced factor VIII or IX activity levels.
Methods
Molecular genetic studies: DNA was extracted from peripheral blood. Coding exons and adjacent intronic regions of F8 and F9 gene were amplified by polymerase chain reaction (PCR) and sequenced on Applied Biosystems® 3500DX Genetic Analyzer. The results were checked in HGMD, ClinVar, dbSNP, gnomAD, and 1000G international databases and different in silico prediction algorithms, such as: PolyPhen-2, SIFT, Mutation Taster, Align GVGD, REVEL and others were consulted.
Results
Three new missense variants were found (table 1). These variants have not been previously reported in the literature or in any of the databases consulted. They do affect the physical-chemical characteristics of the protein (in silico prediction). Therefore, they are classified as likely pathogenic. Because of this, we extended the study to first-degree relatives; finding carriers that were not diagnose at that time. A 20-year-old daughter (patient 1) and a sister (patient 2) received genetic counseling.
Conclusion
These new genetic variants confirm the clinical suspicion of mild hemophilia. The molecular genetic analysis in the family members helps finding asymptomatic carriers so we can provide them genetic counseling. Future reproductive options will be considered in the youngest patient and any family members if needed, firstly and foremostly, prenatal fetal sex chromosome testing by maternal plasma DNA sequencing on female carriers.
Keyword(s): Diagnosis, Gene expression, Hemophilia A, Hemophilia B