

Contributions
Abstract: EP411
Type: E-Poster Presentation
Session title: Acute myeloid leukemia - Biology & Translational Research
Background
Diagnosis of myeloid malignancies requires multiple testing strategies to characterize the genetic makeup of these pathologies. Single nucleotide variants (SNVs), short insertions and deletions (INDELs), copy number variants (CNVs) and other structural variants (inversions and translocations) are the critical genetic abnormalities that dictate therapeutic decisions. While, Next Generation Sequencing (NGS) allows for the detection of all those alterations, only SNV and INDELs detection has been implemented in clinical diagnostic laboratories, limiting the potential of NGS. In fact, CNV detection still relies on karyotype and fluorescence in situ hybridization (FISH) for their detection.
Aims
To develop an algorithm that confidently identifies copy number alterations using a capture-based NGS custom panel currently used only for SNV and INDELs detection.
Methods
We designed a capture-based NGS panel with SOPHiA GENETICS that includes 72 genes with clinical relevance in myeloid neoplasms. The panel includes genes in chromosomes 5, 7, 8, 11, 17 and 20.
The algorithm is based in depth of coverage of the samples, taking into account a unique sample, and comparing it to the whole batch of samples. To generate a batch of normal samples we use 114 samples that have not any previous myeloid neoplasm diagnosis. For the analysis group, we selected a cohort of 19 patients with myeloid malignancies with available NGS data and FISH data for any of the 6 following markers: del(5q), del(7q), +8, amp(11q), del(17p) and del(20q).
Libraries were built according to the SOPHiA GENETICS protocol and sequenced on 600 cycles V3 flow cells in a MiSeq sequencer (Illumina) in batches of 8 samples. The runs were demultiplexed using MiSeq Reporter, and alignment was done by SOPHiA DDM software. Finally, SAMTools was used for depth of coverage calculation and R for custom CNV analysis.
Results
A median of 6.03M reads per sample was obtained, in which 70.13% were on target aligned. We calculated the depth of coverage of those samples and a median of 2835X was obtained. We called every exon on the panel for the copy number.
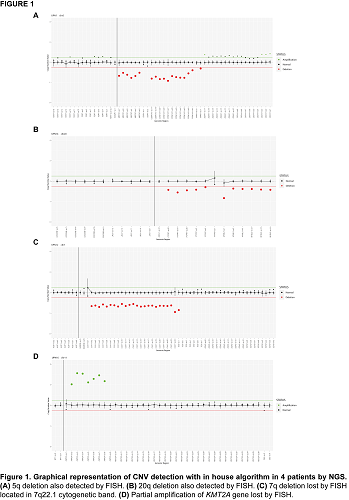
For the 47 FISH results, the concordance was 95.74% (45/47). We detected 8 alterations in 7 patients using NGS approach. We confirmed by NGS the two positive results detected by FISH, a 5q deletion and a 20q deletion (Figure 1A, Figure 1B). In two patients, we detected two alterations in the relevant markers that were not tested by FISH. Additionally, we detected two more alterations in other regions no analyzed by FISH (del(20p) and amp(21q)) in two patients. The two discordant results could be explained taking into account the two different approaches for the analysis.
In UPN12 we detected a 7q deletion by NGS approach (Figure 1C), while it was negative by FISH. This 7q deletion is located in CUX1 gene (7q22.1 cytogenetic band) while our FISH probe is located in 7q31 cytogenetic band.
In UPN14 we detected a partial amplification of KMT2A gene (11q23.3 cytogenetic band) by NGS (Figure 1D), whereas FISH probe was negative. The amplification detected affects exon 2 to exon 8 of the gene, potentially making it undetectable by routine FISH approaches.
Conclusion
Our new designed algorithm allows for the accurate and diagnostic detection of CNV using NGS data. The high concordance between data obtained with our new algorithm and the current gold standard (FISH) demonstrate the feasibility of using NGS data for the detection of CNV in myeloid malignancies. Most importantly, data obtained from NGS offers additional genetic information often overlooked by FISH due to its limitations.
Keyword(s): Cytogenetic abnormalities, FISH, Genomics, Myeloid malignancies
Abstract: EP411
Type: E-Poster Presentation
Session title: Acute myeloid leukemia - Biology & Translational Research
Background
Diagnosis of myeloid malignancies requires multiple testing strategies to characterize the genetic makeup of these pathologies. Single nucleotide variants (SNVs), short insertions and deletions (INDELs), copy number variants (CNVs) and other structural variants (inversions and translocations) are the critical genetic abnormalities that dictate therapeutic decisions. While, Next Generation Sequencing (NGS) allows for the detection of all those alterations, only SNV and INDELs detection has been implemented in clinical diagnostic laboratories, limiting the potential of NGS. In fact, CNV detection still relies on karyotype and fluorescence in situ hybridization (FISH) for their detection.
Aims
To develop an algorithm that confidently identifies copy number alterations using a capture-based NGS custom panel currently used only for SNV and INDELs detection.
Methods
We designed a capture-based NGS panel with SOPHiA GENETICS that includes 72 genes with clinical relevance in myeloid neoplasms. The panel includes genes in chromosomes 5, 7, 8, 11, 17 and 20.
The algorithm is based in depth of coverage of the samples, taking into account a unique sample, and comparing it to the whole batch of samples. To generate a batch of normal samples we use 114 samples that have not any previous myeloid neoplasm diagnosis. For the analysis group, we selected a cohort of 19 patients with myeloid malignancies with available NGS data and FISH data for any of the 6 following markers: del(5q), del(7q), +8, amp(11q), del(17p) and del(20q).
Libraries were built according to the SOPHiA GENETICS protocol and sequenced on 600 cycles V3 flow cells in a MiSeq sequencer (Illumina) in batches of 8 samples. The runs were demultiplexed using MiSeq Reporter, and alignment was done by SOPHiA DDM software. Finally, SAMTools was used for depth of coverage calculation and R for custom CNV analysis.
Results
A median of 6.03M reads per sample was obtained, in which 70.13% were on target aligned. We calculated the depth of coverage of those samples and a median of 2835X was obtained. We called every exon on the panel for the copy number.
For the 47 FISH results, the concordance was 95.74% (45/47). We detected 8 alterations in 7 patients using NGS approach. We confirmed by NGS the two positive results detected by FISH, a 5q deletion and a 20q deletion (Figure 1A, Figure 1B). In two patients, we detected two alterations in the relevant markers that were not tested by FISH. Additionally, we detected two more alterations in other regions no analyzed by FISH (del(20p) and amp(21q)) in two patients. The two discordant results could be explained taking into account the two different approaches for the analysis.
In UPN12 we detected a 7q deletion by NGS approach (Figure 1C), while it was negative by FISH. This 7q deletion is located in CUX1 gene (7q22.1 cytogenetic band) while our FISH probe is located in 7q31 cytogenetic band.
In UPN14 we detected a partial amplification of KMT2A gene (11q23.3 cytogenetic band) by NGS (Figure 1D), whereas FISH probe was negative. The amplification detected affects exon 2 to exon 8 of the gene, potentially making it undetectable by routine FISH approaches.
Conclusion
Our new designed algorithm allows for the accurate and diagnostic detection of CNV using NGS data. The high concordance between data obtained with our new algorithm and the current gold standard (FISH) demonstrate the feasibility of using NGS data for the detection of CNV in myeloid malignancies. Most importantly, data obtained from NGS offers additional genetic information often overlooked by FISH due to its limitations.
Keyword(s): Cytogenetic abnormalities, FISH, Genomics, Myeloid malignancies