

Contributions
Abstract: EP406
Type: E-Poster Presentation
Session title: Acute myeloid leukemia - Biology & Translational Research
Background
Our understanding of how individual mutations, whether present in all or just a fraction of the leukemia cells, affect cellular responses to therapy is limited. Leukemia mouse models provide a unique possibility to explore how therapy affects the evolution of genetically distinct clones and identify mechanisms of resistance allowing transfer to human disease.
Aims
Herein, we studied how different therapies influenced survival, clonal evolution, and resistance patterns in mouse KMT2A-MLLT3 leukemia with subclonal FLT3N676K.
Methods
Bone marrow (BM) from a leukemia expressing KMT2A-MLLT3-mCherry in all cells and a FLT3N676K-GFP in 40% of cells, were re-transplanted to sublethally irradiated recipients. Upon engraftment, treatment was started with either chemotherapy (cytarabine for 5 days (d)+doxorubicin for 3 d), the FLT3 inhibitor AC220, chemotherapy followed by AC220, or AC220+Trametinib, a MEK inhibitor. Targeted treatment was given for 28d; controls received vehicle. The leukemias were analyzed by flow-cytometry, RNA-sequencing and targeted gene re-sequencing.
Results
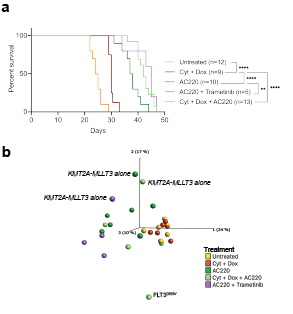
Each treatment prolonged survival with a median latency of 30d for chemotherapy, 37.5d for AC220, 42d for chemotheraphy+AC220, and 45d for AC220+Trametenib, versus 25.5d for the control (Fig 1a). Most cells expressed our marker genes and mice displayed splenomegaly and leukocytosis.
We next determined how treatment impacted evolution of the KMT2A-MLLT3+FLT3N676K cells and while they constituted all cells in control and chemotherapy-treated mice, the other treatments impacted their evolution . Three distinct patterns were discerned with either >80% of KMT2A-MLLT3+FLT3N676K cells, >80% of cells expressing KMT2A-MLLT3 alone, or dual similar sized clones of cells expressing KMT2A-MLLT3 alone or KMT2A-MLLT3+FLT3N676K. Eradication of the FLT3-leukemia cells was rare, but most common in mice receiving AC220+Trametinib and the frequency of dual clones increased when mice received chemotherapy followed by AC220, in line with treatment selectively affecting evolution.
To find clues to treatment resistance, RNA-sequencing (N=44) revealed segregation into three major clusters: 1) leukemias expressing KMT2A-MLLT3 alone, 2) control and chemotherapy-treated leukemias and 3) AC220 treated leukemias. Notably, a set of AC220-treated mice clustered close to the control and chemotherapy-treated mice (Fig 1b). Flow-cytometry data showed that the clustering correlated with the fraction of a myeloid cell population aberrantly expressing B220, which constituted most cells in the control and chemotherapy treated leukemias. Gene set enrichment analysis revealed enrichment of gene sets correlating with stem cells and oxidative phosphorylation, suggesting a switch in cellular phenotype and metabolic state upon resistance in those AC220 treated leukemias. By contrast, the AC220 leukemias in cluster 3, instead showed enrichment of gene sets correlating with granulocyte/macrophage progenitors and immune regulatory pathways, indicating selective dependence of distinct cellular pathways upon resistance. Finally, acquisition of AC220 resistance mutations was rare with a FLT3D835Y and a Ptpn11G503V in two leukemias.
Conclusion
The specific treatment given affected survival and impacted the evolution of genetically distinct cells. The general lack of acquired mutations upon targeted treatment suggests that target-independent mechanisms that result in alternate activation of survival/proliferation explains acquired resistance in a majority of mice.
Keyword(s): AML, Drug resistance, Flt3 inhibitor, MLL
Abstract: EP406
Type: E-Poster Presentation
Session title: Acute myeloid leukemia - Biology & Translational Research
Background
Our understanding of how individual mutations, whether present in all or just a fraction of the leukemia cells, affect cellular responses to therapy is limited. Leukemia mouse models provide a unique possibility to explore how therapy affects the evolution of genetically distinct clones and identify mechanisms of resistance allowing transfer to human disease.
Aims
Herein, we studied how different therapies influenced survival, clonal evolution, and resistance patterns in mouse KMT2A-MLLT3 leukemia with subclonal FLT3N676K.
Methods
Bone marrow (BM) from a leukemia expressing KMT2A-MLLT3-mCherry in all cells and a FLT3N676K-GFP in 40% of cells, were re-transplanted to sublethally irradiated recipients. Upon engraftment, treatment was started with either chemotherapy (cytarabine for 5 days (d)+doxorubicin for 3 d), the FLT3 inhibitor AC220, chemotherapy followed by AC220, or AC220+Trametinib, a MEK inhibitor. Targeted treatment was given for 28d; controls received vehicle. The leukemias were analyzed by flow-cytometry, RNA-sequencing and targeted gene re-sequencing.
Results
Each treatment prolonged survival with a median latency of 30d for chemotherapy, 37.5d for AC220, 42d for chemotheraphy+AC220, and 45d for AC220+Trametenib, versus 25.5d for the control (Fig 1a). Most cells expressed our marker genes and mice displayed splenomegaly and leukocytosis.
We next determined how treatment impacted evolution of the KMT2A-MLLT3+FLT3N676K cells and while they constituted all cells in control and chemotherapy-treated mice, the other treatments impacted their evolution . Three distinct patterns were discerned with either >80% of KMT2A-MLLT3+FLT3N676K cells, >80% of cells expressing KMT2A-MLLT3 alone, or dual similar sized clones of cells expressing KMT2A-MLLT3 alone or KMT2A-MLLT3+FLT3N676K. Eradication of the FLT3-leukemia cells was rare, but most common in mice receiving AC220+Trametinib and the frequency of dual clones increased when mice received chemotherapy followed by AC220, in line with treatment selectively affecting evolution.
To find clues to treatment resistance, RNA-sequencing (N=44) revealed segregation into three major clusters: 1) leukemias expressing KMT2A-MLLT3 alone, 2) control and chemotherapy-treated leukemias and 3) AC220 treated leukemias. Notably, a set of AC220-treated mice clustered close to the control and chemotherapy-treated mice (Fig 1b). Flow-cytometry data showed that the clustering correlated with the fraction of a myeloid cell population aberrantly expressing B220, which constituted most cells in the control and chemotherapy treated leukemias. Gene set enrichment analysis revealed enrichment of gene sets correlating with stem cells and oxidative phosphorylation, suggesting a switch in cellular phenotype and metabolic state upon resistance in those AC220 treated leukemias. By contrast, the AC220 leukemias in cluster 3, instead showed enrichment of gene sets correlating with granulocyte/macrophage progenitors and immune regulatory pathways, indicating selective dependence of distinct cellular pathways upon resistance. Finally, acquisition of AC220 resistance mutations was rare with a FLT3D835Y and a Ptpn11G503V in two leukemias.
Conclusion
The specific treatment given affected survival and impacted the evolution of genetically distinct cells. The general lack of acquired mutations upon targeted treatment suggests that target-independent mechanisms that result in alternate activation of survival/proliferation explains acquired resistance in a majority of mice.
Keyword(s): AML, Drug resistance, Flt3 inhibitor, MLL