

Contributions
Abstract: EP385
Type: E-Poster Presentation
Session title: Acute myeloid leukemia - Biology & Translational Research
Background
Fms-like tyrosine kinase 3 internal tandem duplications (FLT3-ITD) occur in 20-30% of acute myeloid leukemia (AML) cases. These mutations represent both strong prognostic biomarkers and therapeutic targets. Hence, FLT3-ITD are systematically sought and quantified using quantitative fragment analysis, as per European LeukemiaNet guidelines. This method estimates the mutant allele fraction as the ratio of the area under the mutant allele (M) peak divided by the area under the wild type (WT) allele peak. Although robust, this technique has several limitations (high limit of quantification 1%, determination of the size of the ITD, limited application). Next generation sequencing (NGS) may represent an alternative method to detect these mutations with high detection sensitivity and the possibility of sequencing several other genes simultaneously.
However, the use of NGS technologies to detect FLT3-ITD mutations remains challenging because these mutations are heterogeneous in terms of size and/or insertion site, and the perfect or near perfect duplication of the wild-type sequence. These specificities raise new concerns about bio-informatic processes, and several algorithms have been created to detect this duplication (GetITD, km, ScanITD...). However, most of them fail to detect and annotate the broad variety of ITDs. Others are more efficient but must be used in conjunction with manual analysis which is subjective and thus incompatible with routine activity.
Aims
This work aims to develop a kmer-based algorithm named FiLT3r to detect and quantify ITDs and to compare it with existing algorithms to the reference method.
Methods
The different algorithms were tested and compared to the reference method using patients from a multicenter prospective randomized study BIG1 (NCT02416388). This clinical trial included 500 patients aged 18 to 60 years with de novo AML at diagnosis (Acute promyelocytic leukemia and core binding factor AML were excluded from these protocols). Peripheral blood or bone marrow samples from AML patients were screened with our standard NGS routine with a capture method targeting 81 genes (SSQXT Agilent®) and sequenced with Illumina® technology.
Results
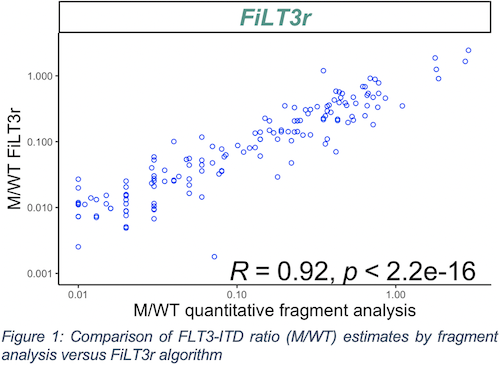
A total of 147 ITDs from 114 FLT3-ITD positive patients (23%) and 71 randomized FLT3-ITD negative patients (14%), determined according to the fragment method, were sequenced (with an average coverage of 2000 paired-end reads on exon 14-15 FLT3). FiLT3r provided results mostly comparable to those obtained by the validated technique, with a sensitivity of 1 and no false positive above a threshold of 1% (limit of quantification of the reference method). All the ITDs detected with fragment analysis of the 114 patients were confirmed using our NGS-based algorithm even in patients with multiple ITDs. The ratios (M/WT) calculated with our algorithm showed a correlation with the reference method estimated at 0.92 (Figure 1) which appears as the highest correlation coefficient of all the algorithms tested (range: 0.42 – 0.92). Among the patients considered negative using fragment analysis, FiLT3r detected five positive cases but these were found at ratios below the quantification threshold of the reference method. Indeed, this algorithm could detect ITD ratios of about 10-4.
Conclusion
FiLT3r can accurately and quickly identify FLT3-ITDs with a robust estimation. These results need further confirmation on a larger cohort but this algorithm represents a promising NGS-based approach to detect FLT3-ITD mutations.
Keyword(s): AML, Flt3-ITD, Molecular markers
Abstract: EP385
Type: E-Poster Presentation
Session title: Acute myeloid leukemia - Biology & Translational Research
Background
Fms-like tyrosine kinase 3 internal tandem duplications (FLT3-ITD) occur in 20-30% of acute myeloid leukemia (AML) cases. These mutations represent both strong prognostic biomarkers and therapeutic targets. Hence, FLT3-ITD are systematically sought and quantified using quantitative fragment analysis, as per European LeukemiaNet guidelines. This method estimates the mutant allele fraction as the ratio of the area under the mutant allele (M) peak divided by the area under the wild type (WT) allele peak. Although robust, this technique has several limitations (high limit of quantification 1%, determination of the size of the ITD, limited application). Next generation sequencing (NGS) may represent an alternative method to detect these mutations with high detection sensitivity and the possibility of sequencing several other genes simultaneously.
However, the use of NGS technologies to detect FLT3-ITD mutations remains challenging because these mutations are heterogeneous in terms of size and/or insertion site, and the perfect or near perfect duplication of the wild-type sequence. These specificities raise new concerns about bio-informatic processes, and several algorithms have been created to detect this duplication (GetITD, km, ScanITD...). However, most of them fail to detect and annotate the broad variety of ITDs. Others are more efficient but must be used in conjunction with manual analysis which is subjective and thus incompatible with routine activity.
Aims
This work aims to develop a kmer-based algorithm named FiLT3r to detect and quantify ITDs and to compare it with existing algorithms to the reference method.
Methods
The different algorithms were tested and compared to the reference method using patients from a multicenter prospective randomized study BIG1 (NCT02416388). This clinical trial included 500 patients aged 18 to 60 years with de novo AML at diagnosis (Acute promyelocytic leukemia and core binding factor AML were excluded from these protocols). Peripheral blood or bone marrow samples from AML patients were screened with our standard NGS routine with a capture method targeting 81 genes (SSQXT Agilent®) and sequenced with Illumina® technology.
Results
A total of 147 ITDs from 114 FLT3-ITD positive patients (23%) and 71 randomized FLT3-ITD negative patients (14%), determined according to the fragment method, were sequenced (with an average coverage of 2000 paired-end reads on exon 14-15 FLT3). FiLT3r provided results mostly comparable to those obtained by the validated technique, with a sensitivity of 1 and no false positive above a threshold of 1% (limit of quantification of the reference method). All the ITDs detected with fragment analysis of the 114 patients were confirmed using our NGS-based algorithm even in patients with multiple ITDs. The ratios (M/WT) calculated with our algorithm showed a correlation with the reference method estimated at 0.92 (Figure 1) which appears as the highest correlation coefficient of all the algorithms tested (range: 0.42 – 0.92). Among the patients considered negative using fragment analysis, FiLT3r detected five positive cases but these were found at ratios below the quantification threshold of the reference method. Indeed, this algorithm could detect ITD ratios of about 10-4.
Conclusion
FiLT3r can accurately and quickly identify FLT3-ITDs with a robust estimation. These results need further confirmation on a larger cohort but this algorithm represents a promising NGS-based approach to detect FLT3-ITD mutations.
Keyword(s): AML, Flt3-ITD, Molecular markers