

Contributions
Abstract: EP375
Type: E-Poster Presentation
Session title: Acute myeloid leukemia - Biology & Translational Research
Background
Acute myeloid leukemia (AML) is a heterogenous disease, characterized by complex patterns of molecular alterations and somatic driver mutations in genes of numerous functional categories, such as NPM1, DNMT3A or IDH1/2. In younger adults, IDH1 mutations occur in 6-10% of AML cases, whereas elderly AML patients show a significantly higher IDH1 mutation frequency of 17.2% accompanied by DNA hypermethylation. Despite advances in targeted therapy due to the recent approval of IDH inhibitors (Enasidenib, Ivosidenib), elderly AML patients with IDH mutations still have an inferior outcome irrespective of cytogenetic group with high rates of chemotherapy resistance and relapses. Recently, the IDH1 p.R132H mutation was linked to altered DNA loop formation by disrupted CTCF binding, leading to oncogene activation in glioma cells, which correlated with inferior outcome.
Aims
Here, we investigate the role of the IDH1 p.R132H mutation in altering 3D DNA architecture and subsequent activation of oncogene expression in AML.
Methods
To identify upregulated oncogenes in IDH1-mutant (-mut) AML, differential gene expression was analyzed in two independent AML datasets, the BeatAML RNA-Seq dataset with n = 264 patients with annotated IDH1-status (Tyner et al. 2018) and a microarray dataset with n = 247 patients (Verhaak et al. 2009). To validate enhanced oncogene expression by RT-qPCR and investigate DNA loop formation, we generated a KG1a-based cellular model containing the IDH1 p.R132H point mutation using CRISPR base editing. Confirmed oncogenes were correlated with differential occupancy and methylation of corresponding CTCF binding sites using chromatin immunoprecipitation (ChIP) and methylation-sensitive restriction followed by RT-qPCR. Clinical relevance of identified oncogenes was investigated by comparing survival of patients with high or low oncogene expression with or without IDH1 mutation in two independent clinical datasets (BeatAML and Bamopoulos et al. 2020, n = 246).
Results
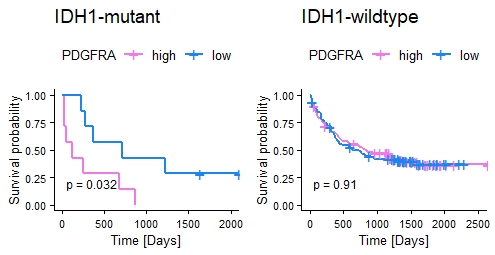
Differential expression analysis of AML datasets identified 179 genes upregulated by >1.5 fold (ANOVA, p < 0.05) in IDH1-mut AML patients compared to IDH1-wildtype (-wt) patients, including 13 oncogenes. Upregulated oncogene expression was confirmed in the KG1a IDH1 p.R132H model revealing highest upregulation (2.5 fold) of the receptor tyrosine kinase gene PDGFRA. Quantification of CTCF occupancy identified decreased CTCF binding by 30% at the CTCF anchor region 1.7 Mb upstream of the PDGFRA locus. In addition, DNA methylation of the identified CTCF binding site was increased by 2-fold in cells harboring the IDH1 p.R132H mutation. Analysis of AML clinical data revealed significantly decreased survival of AML patients with high PDGFRA expression among IDH1-mut but not IDH1-wt patients (Fig. 1, OS of IDH1-mut/-wt patients with high/low PDGFRA, data: Bamopoulos). Interestingly, IDH1-mut KG1a cells showed higher sensitivity towards the tyrosine kinase inhibitor Dasatinib in a WST-based drug screen, supporting Dasatinib as novel candidate for treatment of AML patients with IDH1 mutation.
Conclusion
Our data suggest that IDH1 p.R132H mutation leads to 3D conformational changes in AML, disrupting DNA loop formation and insulation of the tyrosine kinase PDGFRA, which subsequently results in upregulated PDGFRA oncogene expression. As high PDGFRA expression correlated with poor prognosis, treatment with tyrosine kinase inhibitor Dasatinib may be an option for IDH1 p.R132H mutant AML patients. Our findings offer a base for novel treatment strategies and tackling resistance in AML patients with IDH1 mutations.
Keyword(s): AML, Chromatin structure, PDGFRA, Tyrosine kinase inhibitor
Abstract: EP375
Type: E-Poster Presentation
Session title: Acute myeloid leukemia - Biology & Translational Research
Background
Acute myeloid leukemia (AML) is a heterogenous disease, characterized by complex patterns of molecular alterations and somatic driver mutations in genes of numerous functional categories, such as NPM1, DNMT3A or IDH1/2. In younger adults, IDH1 mutations occur in 6-10% of AML cases, whereas elderly AML patients show a significantly higher IDH1 mutation frequency of 17.2% accompanied by DNA hypermethylation. Despite advances in targeted therapy due to the recent approval of IDH inhibitors (Enasidenib, Ivosidenib), elderly AML patients with IDH mutations still have an inferior outcome irrespective of cytogenetic group with high rates of chemotherapy resistance and relapses. Recently, the IDH1 p.R132H mutation was linked to altered DNA loop formation by disrupted CTCF binding, leading to oncogene activation in glioma cells, which correlated with inferior outcome.
Aims
Here, we investigate the role of the IDH1 p.R132H mutation in altering 3D DNA architecture and subsequent activation of oncogene expression in AML.
Methods
To identify upregulated oncogenes in IDH1-mutant (-mut) AML, differential gene expression was analyzed in two independent AML datasets, the BeatAML RNA-Seq dataset with n = 264 patients with annotated IDH1-status (Tyner et al. 2018) and a microarray dataset with n = 247 patients (Verhaak et al. 2009). To validate enhanced oncogene expression by RT-qPCR and investigate DNA loop formation, we generated a KG1a-based cellular model containing the IDH1 p.R132H point mutation using CRISPR base editing. Confirmed oncogenes were correlated with differential occupancy and methylation of corresponding CTCF binding sites using chromatin immunoprecipitation (ChIP) and methylation-sensitive restriction followed by RT-qPCR. Clinical relevance of identified oncogenes was investigated by comparing survival of patients with high or low oncogene expression with or without IDH1 mutation in two independent clinical datasets (BeatAML and Bamopoulos et al. 2020, n = 246).
Results
Differential expression analysis of AML datasets identified 179 genes upregulated by >1.5 fold (ANOVA, p < 0.05) in IDH1-mut AML patients compared to IDH1-wildtype (-wt) patients, including 13 oncogenes. Upregulated oncogene expression was confirmed in the KG1a IDH1 p.R132H model revealing highest upregulation (2.5 fold) of the receptor tyrosine kinase gene PDGFRA. Quantification of CTCF occupancy identified decreased CTCF binding by 30% at the CTCF anchor region 1.7 Mb upstream of the PDGFRA locus. In addition, DNA methylation of the identified CTCF binding site was increased by 2-fold in cells harboring the IDH1 p.R132H mutation. Analysis of AML clinical data revealed significantly decreased survival of AML patients with high PDGFRA expression among IDH1-mut but not IDH1-wt patients (Fig. 1, OS of IDH1-mut/-wt patients with high/low PDGFRA, data: Bamopoulos). Interestingly, IDH1-mut KG1a cells showed higher sensitivity towards the tyrosine kinase inhibitor Dasatinib in a WST-based drug screen, supporting Dasatinib as novel candidate for treatment of AML patients with IDH1 mutation.
Conclusion
Our data suggest that IDH1 p.R132H mutation leads to 3D conformational changes in AML, disrupting DNA loop formation and insulation of the tyrosine kinase PDGFRA, which subsequently results in upregulated PDGFRA oncogene expression. As high PDGFRA expression correlated with poor prognosis, treatment with tyrosine kinase inhibitor Dasatinib may be an option for IDH1 p.R132H mutant AML patients. Our findings offer a base for novel treatment strategies and tackling resistance in AML patients with IDH1 mutations.
Keyword(s): AML, Chromatin structure, PDGFRA, Tyrosine kinase inhibitor