

Contributions
Abstract: EP332
Type: E-Poster Presentation
Session title: Acute lymphoblastic leukemia - Biology & Translational Research
Background
Philadelphia positive acute lymphoblastic leukaemia (Ph+ ALL) is a high risk ALL subtype with an aggressive clinical course. While the incorporation of tyrosine kinase inhibitors (TKIs) into the chemotherapy regimen has improved upfront treatment, the development of resistance/relapse to TKIs is still a major problem. RAS pathway mutations (PTPN11, NRAS and KRAS) are prevalent in relapsed B-ALL and confer poor prognosis. Therefore, new targeted therapies to overcome the resistance induced by these mutations are needed.
Aims
To investigate targeted therapies to overcome the resistance induced by activating lesions in the RAS pathway in cell line models of Ph+ ALL.
Methods
Three Ph+ ALL SUP-B15 (p190 BCR-ABL1) resistant cell lines were generated by long term dose escalation of imatinib (to 5 µM, SUP-B15 IR), dasatinib (to 200 nM, DR) and ponatinib (to 200 nM, PR). Drug sensitivity was measured by Annexin V and viability stain 780. RNAseq and western blot analysis were performed to identify mechanisms of resistance. The combination index (CI) was calculated by Calcusyn software.
Results
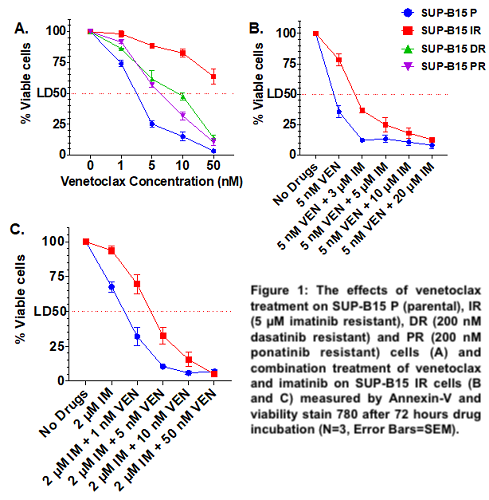
In comparison to parental, the SUP-B15 IR, DR and PR cell lines were resistant to all TKIs (imatinib, nilotinib, dasatinib and ponatinib) and to asciminib. Interestingly, SUP-B15 IR showed primary resistance to venetoclax (VEN) (LD50 = 74.94 nM vs 2.1 nM in the parental, p=0.0054), while DR and PR cell lines were sensitive (LD50 = 8.36 nM and 5.98 nM respectively) (Figure 1A). RNAseq analysis identified KRAS p.G12S (VAF 37%) and NRAS p.G12S (VAF 100%) mutations in the DR and PR cell lines respectively. Two PTPN11 (p.A461T, VAF 63.7% and p.P491H, VAF 40.2%) mutations were identified in the catalytic domain of tyrosine-protein phosphatase in SUP-B15 IR cells. Activating lesions in PTPN11 cause an oncogenic ERK signalling, which stabilises the anti-apoptotic protein Mcl-1 and is the likely cause of venetoclax resistance in acute myeloid leukaemia. We therefore investigated the activation of the MAPK/ERK pathway and Mcl-1 by western blot. SUPB15 IRPTPN11mut had increased expression of pBCR-ABL (P=0.002), pERK1/2 (P<0.0001), Mcl-1 (P<0.0001) and similar levels of Bcl-2 in comparison to the sensitive cell lines. Because treatment with imatinib alone (0, 2, 5 and 10µM) led to a dose-dependent reduction of pBCR-ABL1 (Y177), pERK1/2 and Mcl-1 without affecting Bcl-2, we investigated whether co-targeting Mcl-1 and Bcl-2 could be an effective strategy for inducing cell death in SUPB15 IRPTPN11 mut cell lines. Whilst these cell lines were resistant to the Mcl-1‐selective inhibitor S63845 (LD50=290 nM vs 78.5 nM for parental), combination therapy with venetoclax was synergistic in overcoming resistance (CI=0.051 for 50 nM S63845 and 5 nM venetoclax). Interestingly, combination treatment with imatinib and venetoclax (CI = 0.075 for 2 µM IM and 5 nM VEN) (Figure 1B and 1C) or asciminib and venetoclax (CI=0.032 for 2 µM asciminib and 5 nM venetoclax) was also effective in inducing cell death. This combination therapy significantly reduced pERK1/2 (P<0.0001) and Mcl-1 expression (P<0.0001) and increased cleaved-PARP level (P=0.024).
The DRKRASmut and PRNRASmut lines instead showed overexpression of Bcl-2 (P=0.0079 and P=0.01 respectively), but not pBCR-ABL or Mcl-1, which could explain why venetoclax alone overcame resistance in these cell lines
Conclusion
Combination therapy of TKIs and venetoclax could be promising for Ph+ ALL patients carrying PTPN11 mutations and Mcl-1 overexpression. Studies are ongoing to determine the role of the mutations in PTPN11 in the development of the resistance.
Keyword(s): Ph+ ALL, PTPN11, Resistance, Targeted therapy
Abstract: EP332
Type: E-Poster Presentation
Session title: Acute lymphoblastic leukemia - Biology & Translational Research
Background
Philadelphia positive acute lymphoblastic leukaemia (Ph+ ALL) is a high risk ALL subtype with an aggressive clinical course. While the incorporation of tyrosine kinase inhibitors (TKIs) into the chemotherapy regimen has improved upfront treatment, the development of resistance/relapse to TKIs is still a major problem. RAS pathway mutations (PTPN11, NRAS and KRAS) are prevalent in relapsed B-ALL and confer poor prognosis. Therefore, new targeted therapies to overcome the resistance induced by these mutations are needed.
Aims
To investigate targeted therapies to overcome the resistance induced by activating lesions in the RAS pathway in cell line models of Ph+ ALL.
Methods
Three Ph+ ALL SUP-B15 (p190 BCR-ABL1) resistant cell lines were generated by long term dose escalation of imatinib (to 5 µM, SUP-B15 IR), dasatinib (to 200 nM, DR) and ponatinib (to 200 nM, PR). Drug sensitivity was measured by Annexin V and viability stain 780. RNAseq and western blot analysis were performed to identify mechanisms of resistance. The combination index (CI) was calculated by Calcusyn software.
Results
In comparison to parental, the SUP-B15 IR, DR and PR cell lines were resistant to all TKIs (imatinib, nilotinib, dasatinib and ponatinib) and to asciminib. Interestingly, SUP-B15 IR showed primary resistance to venetoclax (VEN) (LD50 = 74.94 nM vs 2.1 nM in the parental, p=0.0054), while DR and PR cell lines were sensitive (LD50 = 8.36 nM and 5.98 nM respectively) (Figure 1A). RNAseq analysis identified KRAS p.G12S (VAF 37%) and NRAS p.G12S (VAF 100%) mutations in the DR and PR cell lines respectively. Two PTPN11 (p.A461T, VAF 63.7% and p.P491H, VAF 40.2%) mutations were identified in the catalytic domain of tyrosine-protein phosphatase in SUP-B15 IR cells. Activating lesions in PTPN11 cause an oncogenic ERK signalling, which stabilises the anti-apoptotic protein Mcl-1 and is the likely cause of venetoclax resistance in acute myeloid leukaemia. We therefore investigated the activation of the MAPK/ERK pathway and Mcl-1 by western blot. SUPB15 IRPTPN11mut had increased expression of pBCR-ABL (P=0.002), pERK1/2 (P<0.0001), Mcl-1 (P<0.0001) and similar levels of Bcl-2 in comparison to the sensitive cell lines. Because treatment with imatinib alone (0, 2, 5 and 10µM) led to a dose-dependent reduction of pBCR-ABL1 (Y177), pERK1/2 and Mcl-1 without affecting Bcl-2, we investigated whether co-targeting Mcl-1 and Bcl-2 could be an effective strategy for inducing cell death in SUPB15 IRPTPN11 mut cell lines. Whilst these cell lines were resistant to the Mcl-1‐selective inhibitor S63845 (LD50=290 nM vs 78.5 nM for parental), combination therapy with venetoclax was synergistic in overcoming resistance (CI=0.051 for 50 nM S63845 and 5 nM venetoclax). Interestingly, combination treatment with imatinib and venetoclax (CI = 0.075 for 2 µM IM and 5 nM VEN) (Figure 1B and 1C) or asciminib and venetoclax (CI=0.032 for 2 µM asciminib and 5 nM venetoclax) was also effective in inducing cell death. This combination therapy significantly reduced pERK1/2 (P<0.0001) and Mcl-1 expression (P<0.0001) and increased cleaved-PARP level (P=0.024).
The DRKRASmut and PRNRASmut lines instead showed overexpression of Bcl-2 (P=0.0079 and P=0.01 respectively), but not pBCR-ABL or Mcl-1, which could explain why venetoclax alone overcame resistance in these cell lines
Conclusion
Combination therapy of TKIs and venetoclax could be promising for Ph+ ALL patients carrying PTPN11 mutations and Mcl-1 overexpression. Studies are ongoing to determine the role of the mutations in PTPN11 in the development of the resistance.
Keyword(s): Ph+ ALL, PTPN11, Resistance, Targeted therapy