

Contributions
Abstract: EP1301
Type: E-Poster Presentation
Session title: Thalassemias
Background
Phase 3 studies evaluating betibeglogene autotemcel (beti-cel; LentiGlobin for β-thalassemia) gene therapy in patients with TDT, HGB-207 (non-β0/β0 genotypes; NCT02906202) and HGB-212 (β0/β0, β0/β+IVS-I-110, β+IVS-I-110/β+IVS-I-110 genotypes; NCT03207009), demonstrated positive outcomes in adults.
Aims
Therefore, the studies were expanded to include patients <18 years of age. We report interim results in these pediatric patients.
Methods
Autologous CD34+ cells were transduced ex vivo with BB305 lentiviral vector to produce beti-cel. Patients underwent pharmacokinetic-adjusted busulfan-based myeloablation followed by beti-cel infusion. Transfusion independence (TI; weighted average hemoglobin [Hb] ≥9 g/dL without packed red blood cell transfusions for ≥12 months) was the HGB-207 and HGB-212 (as of protocol amendment dated Oct 8, 2020) primary endpoint. Data are presented as median (min–max).
Results
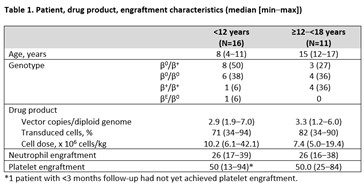
As of 30 November 2020, 27 pediatric patients were treated and followed for 23.1 (0.9–35.5) months, including 16 patients <12 years (HGB-207: n=8; HGB-212: n=8) and 11 patients ≥12‒<18 years at assent (HGB-207: n=6; HGB-212: n=5). The youngest patients were 4 years of age (n=3). TI was achieved in 9/11 evaluable patients <12 years and 10/10 evaluable patients ≥12–<18 years. The median duration of ongoing TI was 19.5 (12.3–32.0) months. Pediatric patients achieved TI at a similar rate to adults (90.5% [19/21] vs 84.6% [11/13] in patients > 18 years).
Weighted average Hb during TI in patients <12 years and ≥12–<18 years was 10.0 (9.5‒11.4) g/dL and 11.5 (9.6‒13.0) g/dL, respectively. At last visit, gene therapy-derived HbAT87Q levels in these patients were 8.4 (5.0–10.1) g/dL (n=9) and 9.1 (4.4–10.8) g/dL (n=10), respectively. These data are comparable to those recorded in patients ≥18 years who achieved TI; weighted average Hb was 12.6 (9.9 – 13.6) g/dL and HbAT87Q at last visit was 9.9 (6.3 – 13.4) g/dL (n=11). The median nadir lymphocytes within the first month post-infusion for patients <18 years was 0.48 x109/L (min – max: 0.02 – 1.7 x109/L) (n=27).
Non-hematologic ≥ Grade 3 adverse events in ≥3 patients <18 years were stomatitis (n=14), febrile neutropenia (n=13), epistaxis (n=6), decreased appetite (n=5), increased alanine aminotransferase (n=3), hypoxia (n=3), and pyrexia (n=3). Grade 4 veno-occlusive liver disease occurred in 2 patients ≥12–<18 years and one grade 2 event occurred in a patient <12 years; all events resolved after treatment with defibrotide. Drug-product related AEs as per investigators opinion were reported in 2 patients < 12 years of age (thrombocytopenia, tachycardia) and 2 patients 12-18 years of age (abdominal pain (2)). No replication-competent lentivirus was reported. All patients had polyclonal vector integration; no integration site contributed >3% of all integration sites at last assessment (n=22).
Conclusion
Pediatric patients with diverse TDT genotypes achieved TI rates comparable to adults after beti-cel gene therapy, suggesting that beti-cel is a viable treatment option for patients with TDT of all ages. The treatment regimen had a safety profile consistent with busulfan myeloablation.
Keyword(s):
Abstract: EP1301
Type: E-Poster Presentation
Session title: Thalassemias
Background
Phase 3 studies evaluating betibeglogene autotemcel (beti-cel; LentiGlobin for β-thalassemia) gene therapy in patients with TDT, HGB-207 (non-β0/β0 genotypes; NCT02906202) and HGB-212 (β0/β0, β0/β+IVS-I-110, β+IVS-I-110/β+IVS-I-110 genotypes; NCT03207009), demonstrated positive outcomes in adults.
Aims
Therefore, the studies were expanded to include patients <18 years of age. We report interim results in these pediatric patients.
Methods
Autologous CD34+ cells were transduced ex vivo with BB305 lentiviral vector to produce beti-cel. Patients underwent pharmacokinetic-adjusted busulfan-based myeloablation followed by beti-cel infusion. Transfusion independence (TI; weighted average hemoglobin [Hb] ≥9 g/dL without packed red blood cell transfusions for ≥12 months) was the HGB-207 and HGB-212 (as of protocol amendment dated Oct 8, 2020) primary endpoint. Data are presented as median (min–max).
Results
As of 30 November 2020, 27 pediatric patients were treated and followed for 23.1 (0.9–35.5) months, including 16 patients <12 years (HGB-207: n=8; HGB-212: n=8) and 11 patients ≥12‒<18 years at assent (HGB-207: n=6; HGB-212: n=5). The youngest patients were 4 years of age (n=3). TI was achieved in 9/11 evaluable patients <12 years and 10/10 evaluable patients ≥12–<18 years. The median duration of ongoing TI was 19.5 (12.3–32.0) months. Pediatric patients achieved TI at a similar rate to adults (90.5% [19/21] vs 84.6% [11/13] in patients > 18 years).
Weighted average Hb during TI in patients <12 years and ≥12–<18 years was 10.0 (9.5‒11.4) g/dL and 11.5 (9.6‒13.0) g/dL, respectively. At last visit, gene therapy-derived HbAT87Q levels in these patients were 8.4 (5.0–10.1) g/dL (n=9) and 9.1 (4.4–10.8) g/dL (n=10), respectively. These data are comparable to those recorded in patients ≥18 years who achieved TI; weighted average Hb was 12.6 (9.9 – 13.6) g/dL and HbAT87Q at last visit was 9.9 (6.3 – 13.4) g/dL (n=11). The median nadir lymphocytes within the first month post-infusion for patients <18 years was 0.48 x109/L (min – max: 0.02 – 1.7 x109/L) (n=27).
Non-hematologic ≥ Grade 3 adverse events in ≥3 patients <18 years were stomatitis (n=14), febrile neutropenia (n=13), epistaxis (n=6), decreased appetite (n=5), increased alanine aminotransferase (n=3), hypoxia (n=3), and pyrexia (n=3). Grade 4 veno-occlusive liver disease occurred in 2 patients ≥12–<18 years and one grade 2 event occurred in a patient <12 years; all events resolved after treatment with defibrotide. Drug-product related AEs as per investigators opinion were reported in 2 patients < 12 years of age (thrombocytopenia, tachycardia) and 2 patients 12-18 years of age (abdominal pain (2)). No replication-competent lentivirus was reported. All patients had polyclonal vector integration; no integration site contributed >3% of all integration sites at last assessment (n=22).
Conclusion
Pediatric patients with diverse TDT genotypes achieved TI rates comparable to adults after beti-cel gene therapy, suggesting that beti-cel is a viable treatment option for patients with TDT of all ages. The treatment regimen had a safety profile consistent with busulfan myeloablation.
Keyword(s):