

Contributions
Abstract: EP1212
Type: E-Poster Presentation
Session title: Sickle cell disease
Background
Hydroxyurea (HU) and chronic transfusion therapy (CTT) are the only treatments currently available in the UK to modify the course of sickle cell disease (SCD), while haematopoietic stem cell transplant (HSCT) is only available to a small minority with HLA-matched sibling donor. Numerous studies have shown reduction in acute vaso-occlusive and neurological complications (particularly ischaemic stroke) with these therapies. New therapies under development are expected to further improve outcomes, but there remains uncertainty towards the degree to which morbidity and mortality could be improved and how to evaluate their health economic potential. There is, therefore, a need for accurate, long-term follow-up studies demonstrating the range of complications, residual morbidity and early mortality. The East London Newborn Cohort Study aims to study long-term outcomes of patients with SCD in the UK. Outcomes have previously been reported up to the end of 2005 (Telfer et al, Haematologica, 2007). Here, we report on follow-up during the years 2015-2018 when HU and CTT were widely available to children and adults according to national and international guidelines.
Aims
To compare impact of current standard therapies on rates of acute complications.
Methods
Eligible patients were born in a designated local region, diagnosed by newborn screening between 1983-2018 and followed in our Specialist Centre. The age distribution in this cohort is currently paediatric and young adult. Recruitment was at birth, before any SCD complications could occur and represent the full spectrum of disease severity, avoiding any selection bias from clinical phenotype. For this study, we restricted the analysis to children and adults with HbSS and HbS beta thalassaemia who were followed in our centre during the period 2015-2018. Patients were categorised according to treatment received during follow-up (HU, CTT or no treatment), where duration was at least 6 months. Patients who switched were allocated according to the longest duration of therapy in follow-up. Data on all acute complications requiring hospital treatment (in patient, emergency department, or day unit) were collected. Sickle cell complications included acute (pain, chest syndrome, ischaemic stroke, hepatic/splenic sequestration), aplastic crisis, dactylitis, girdle syndrome, and priapism. Chi-squared tests were used to test for associations between categorical variables, and the Kruskal Wallis test for comparisons of continuous variables between the three treatment groups.
Results
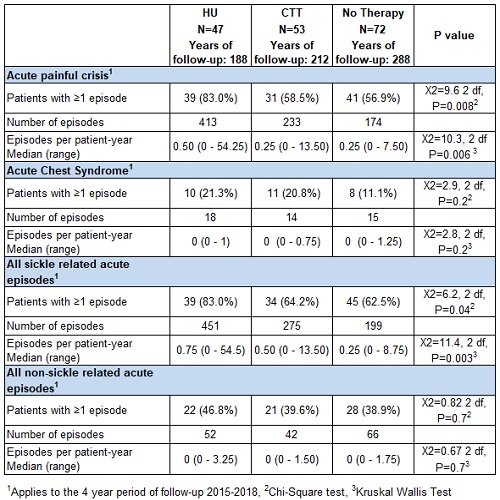
175 patients were included; 91 (52.0%) were female. Median age at study entry was 11.6 years (range 0.2 to 31.4 years). 47 (26.9%) patients were treated with HU, 53 (30.8%) with CTT, 72 (41.9%) were not treated, 2 (1.1%) died, and 1 (0.6%) received HSCT. There was a total of 695 patient-years of follow-up. Data on acute complications are presented in the table. More than one-third of the patients had a relatively mild phenotype and had not been treated with HU or CTT. This group had the lowest rate of acute SCD-related complications, while patients on HU had the highest rates of pain, acute chest syndrome, and sickle complications.
Conclusion
The results confirm that current standard therapies are only partially effective in controlling acute complications of SCD. There remains a significant unmet need for effective disease modifying or curative therapies for patients with more severe phenotype.
Keyword(s): Blood transfusion, Clinical data, Hydroxyurea, Sickle cell patient
Abstract: EP1212
Type: E-Poster Presentation
Session title: Sickle cell disease
Background
Hydroxyurea (HU) and chronic transfusion therapy (CTT) are the only treatments currently available in the UK to modify the course of sickle cell disease (SCD), while haematopoietic stem cell transplant (HSCT) is only available to a small minority with HLA-matched sibling donor. Numerous studies have shown reduction in acute vaso-occlusive and neurological complications (particularly ischaemic stroke) with these therapies. New therapies under development are expected to further improve outcomes, but there remains uncertainty towards the degree to which morbidity and mortality could be improved and how to evaluate their health economic potential. There is, therefore, a need for accurate, long-term follow-up studies demonstrating the range of complications, residual morbidity and early mortality. The East London Newborn Cohort Study aims to study long-term outcomes of patients with SCD in the UK. Outcomes have previously been reported up to the end of 2005 (Telfer et al, Haematologica, 2007). Here, we report on follow-up during the years 2015-2018 when HU and CTT were widely available to children and adults according to national and international guidelines.
Aims
To compare impact of current standard therapies on rates of acute complications.
Methods
Eligible patients were born in a designated local region, diagnosed by newborn screening between 1983-2018 and followed in our Specialist Centre. The age distribution in this cohort is currently paediatric and young adult. Recruitment was at birth, before any SCD complications could occur and represent the full spectrum of disease severity, avoiding any selection bias from clinical phenotype. For this study, we restricted the analysis to children and adults with HbSS and HbS beta thalassaemia who were followed in our centre during the period 2015-2018. Patients were categorised according to treatment received during follow-up (HU, CTT or no treatment), where duration was at least 6 months. Patients who switched were allocated according to the longest duration of therapy in follow-up. Data on all acute complications requiring hospital treatment (in patient, emergency department, or day unit) were collected. Sickle cell complications included acute (pain, chest syndrome, ischaemic stroke, hepatic/splenic sequestration), aplastic crisis, dactylitis, girdle syndrome, and priapism. Chi-squared tests were used to test for associations between categorical variables, and the Kruskal Wallis test for comparisons of continuous variables between the three treatment groups.
Results
175 patients were included; 91 (52.0%) were female. Median age at study entry was 11.6 years (range 0.2 to 31.4 years). 47 (26.9%) patients were treated with HU, 53 (30.8%) with CTT, 72 (41.9%) were not treated, 2 (1.1%) died, and 1 (0.6%) received HSCT. There was a total of 695 patient-years of follow-up. Data on acute complications are presented in the table. More than one-third of the patients had a relatively mild phenotype and had not been treated with HU or CTT. This group had the lowest rate of acute SCD-related complications, while patients on HU had the highest rates of pain, acute chest syndrome, and sickle complications.
Conclusion
The results confirm that current standard therapies are only partially effective in controlling acute complications of SCD. There remains a significant unmet need for effective disease modifying or curative therapies for patients with more severe phenotype.
Keyword(s): Blood transfusion, Clinical data, Hydroxyurea, Sickle cell patient