

Contributions
Abstract: EP1141
Type: E-Poster Presentation
Session title: Platelet disorders
Background
Evans syndrome (ES) is a rare condition, defined as the presence of two autoimmune cytopenias (AIC), more frequently autoimmune hemolytic anemia (AIHA) and immune thrombocytopenia (ITP), and rarely autoimmune neutropenia (AIN). ES can be classified as primary or secondary to various conditions, including lymphoproliferative disease (LPD), primary immune deficiencies (PID) or other systemic autoimmune diseases (AID). ES onset may be acute and life-threatening, whilst its clinical course is usually chronic and marked by several relapses of AIHA, ITP or both. First line therapy is based on steroids +/- intravenous immunoglobulins (IVIG), followed by rituximab and splenectomy in refractory/relapsing cases. Concerning ITP, splenectomy is not always feasible, and rituximab is poorly effective on the long term. Thrombopoietin receptors agonists (TPO-RA), such as romiplostim (ROMI) and eltrombopag (EPAG), are highly effective in primary ITP, whilst their use in ES has never been systematically studied.
Aims
to evaluate the efficacy and safety of TPO-RA in a cohort of patients with ES.
Methods
ES patients treated with TPO-RA out of a cohort of 46 cases followed at a tertiary hematologic hospital were retrospectively evaluated. Baseline hematologic parameters, associated conditions, previous/concomitant treatments were assessed. The time from diagnosis to first TPO-RA was collected. Response rates were evaluated at 1, 3, 6, and 12 months, and classified as partial (PR) or complete (CR), for platelets >30x10^9/L or >100x10^9/L, respectively. Treatment emergent adverse events (TEAEs) were registered and graded according to CTCAE.
Results
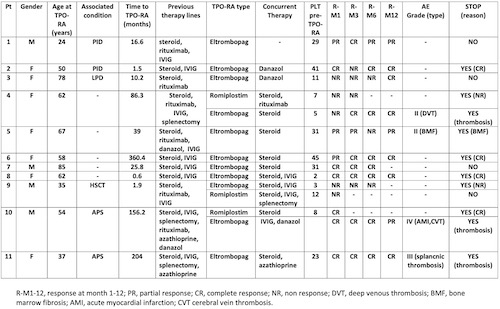
As shown in Table 1, 11 patients have been evaluated: 4 males and 6 females, with a median age at the start of TPO-RA of 55.6 years (24-85). Six patients suffered from secondary ES: 2 PID, 2 antiphospholipid syndrome (APS), 1 post allogenic stem cells transplant (SCT), and 1 LPD. All patients had received steroids +/- IVIG, and the majority at least one further line including rituximab (N=6), splenectomy (N=3), azathioprine (N=2), and danazol (N=1). The median time from diagnosis to TPO-RA was 25.8 months (0.6-360.4). Response rates to the first TPO-RA (9 EPAG and 2 ROMI) were: 72% at month 1 (45% CR, 27% PR, N=11), 60% at month 3 (50% CR, 10% PR, N=10); 66% at month 6 (55% CR 11% PR, N=9); 100% at 12 months (71% CR, 28% PR, N=7). Of note, 91% of patients required concomitant therapies, including steroids +/- IVIG (N=10), danazol (N=3), rituximab, splenectomy and azathioprine (N=1 each). Interestingly, 3 patients switched to the alternative TPO-RA (2 ROMI to EPAG and 1 vice versa) mainly because of no response (NR); two out of 3 responded. Four out of 11 (36%) patients developed at least one TEAE: 3 venous thromboses (1 deep venous thrombosis, 1 cerebral vein thrombosis CVT, and 1 splanchnic thrombosis) and 1 acute myocardial infarction (in the same APS patient experiencing CVT). Four patients are still receiving TPO-RA, whilst the other stopped because of persistent CR (N=3), thrombosis (N=3), or increase in bone marrow reticulin fibrosis (N=1).
Conclusion
TPO-RA were effective in about 70% of ES patients, even heavily pre-treated, but required a concomitant therapy in the majority of them. Moreover, TPO-RA use was complicated by a high occurrence of thrombotic events that may be also favored by underlying conditions. These findings highlight that ES is sustained by a profound immune dysregulation where autoimmune platelet destruction cannot be completely overcome by bone marrow stimulation.
Keyword(s): Autoimmune hemolytic anemia (AIHA), Immune thrombocytopenia (ITP), Thrombopoietin (TPO), Thrombosis
Abstract: EP1141
Type: E-Poster Presentation
Session title: Platelet disorders
Background
Evans syndrome (ES) is a rare condition, defined as the presence of two autoimmune cytopenias (AIC), more frequently autoimmune hemolytic anemia (AIHA) and immune thrombocytopenia (ITP), and rarely autoimmune neutropenia (AIN). ES can be classified as primary or secondary to various conditions, including lymphoproliferative disease (LPD), primary immune deficiencies (PID) or other systemic autoimmune diseases (AID). ES onset may be acute and life-threatening, whilst its clinical course is usually chronic and marked by several relapses of AIHA, ITP or both. First line therapy is based on steroids +/- intravenous immunoglobulins (IVIG), followed by rituximab and splenectomy in refractory/relapsing cases. Concerning ITP, splenectomy is not always feasible, and rituximab is poorly effective on the long term. Thrombopoietin receptors agonists (TPO-RA), such as romiplostim (ROMI) and eltrombopag (EPAG), are highly effective in primary ITP, whilst their use in ES has never been systematically studied.
Aims
to evaluate the efficacy and safety of TPO-RA in a cohort of patients with ES.
Methods
ES patients treated with TPO-RA out of a cohort of 46 cases followed at a tertiary hematologic hospital were retrospectively evaluated. Baseline hematologic parameters, associated conditions, previous/concomitant treatments were assessed. The time from diagnosis to first TPO-RA was collected. Response rates were evaluated at 1, 3, 6, and 12 months, and classified as partial (PR) or complete (CR), for platelets >30x10^9/L or >100x10^9/L, respectively. Treatment emergent adverse events (TEAEs) were registered and graded according to CTCAE.
Results
As shown in Table 1, 11 patients have been evaluated: 4 males and 6 females, with a median age at the start of TPO-RA of 55.6 years (24-85). Six patients suffered from secondary ES: 2 PID, 2 antiphospholipid syndrome (APS), 1 post allogenic stem cells transplant (SCT), and 1 LPD. All patients had received steroids +/- IVIG, and the majority at least one further line including rituximab (N=6), splenectomy (N=3), azathioprine (N=2), and danazol (N=1). The median time from diagnosis to TPO-RA was 25.8 months (0.6-360.4). Response rates to the first TPO-RA (9 EPAG and 2 ROMI) were: 72% at month 1 (45% CR, 27% PR, N=11), 60% at month 3 (50% CR, 10% PR, N=10); 66% at month 6 (55% CR 11% PR, N=9); 100% at 12 months (71% CR, 28% PR, N=7). Of note, 91% of patients required concomitant therapies, including steroids +/- IVIG (N=10), danazol (N=3), rituximab, splenectomy and azathioprine (N=1 each). Interestingly, 3 patients switched to the alternative TPO-RA (2 ROMI to EPAG and 1 vice versa) mainly because of no response (NR); two out of 3 responded. Four out of 11 (36%) patients developed at least one TEAE: 3 venous thromboses (1 deep venous thrombosis, 1 cerebral vein thrombosis CVT, and 1 splanchnic thrombosis) and 1 acute myocardial infarction (in the same APS patient experiencing CVT). Four patients are still receiving TPO-RA, whilst the other stopped because of persistent CR (N=3), thrombosis (N=3), or increase in bone marrow reticulin fibrosis (N=1).
Conclusion
TPO-RA were effective in about 70% of ES patients, even heavily pre-treated, but required a concomitant therapy in the majority of them. Moreover, TPO-RA use was complicated by a high occurrence of thrombotic events that may be also favored by underlying conditions. These findings highlight that ES is sustained by a profound immune dysregulation where autoimmune platelet destruction cannot be completely overcome by bone marrow stimulation.
Keyword(s): Autoimmune hemolytic anemia (AIHA), Immune thrombocytopenia (ITP), Thrombopoietin (TPO), Thrombosis