

Contributions
Abstract: EP1116
Type: E-Poster Presentation
Session title: Myeloproliferative neoplasms - Clinical
Background
The coexistence of myeloproliferative neoplasms (MPN) and lymphoproliferative neoplasms (LPN) is a rare finding. However, studies have demonstrated that patients with MPN have approximately a 3-fold higher risk of developing an LPN than the general population. The clonal origin of these diseases has yet to be elucidated, although there are suggestions that their origin may lie in multipotent hematopoietic stem cells.
Aims
Use of next-generation sequencing (NGS) to identify myeloid mutations in separated cellular populations associated with the development of coexisting MPN/LPN syndromes.
Methods
All patients with a MPN or LPN diagnosis (dx) between 1997 and 2020 were retrospectively analyzed for concomitant MPN/LPN according to the WHO 2016 criteria. Of the 21 patients identified, informed consent was obtained from the live patients (n=13) for peripheral blood (PB) extraction. CD34+ (progenitor cells), CD15+ (granulocytes), and CD19+ (B lymphocytes) cell populations were separated using an autoMACS Pro. So far, genomic DNA of 5 patients has been analyzed by NGS with the MiSeq (Illumina) platform using the targeted panel Myeloid Solution™ (SOPHiA GENETICS). Only variants with VAF ≥1%, MAF<1% and described as having a pathogenic or probably pathogenic effect were considered.
Results
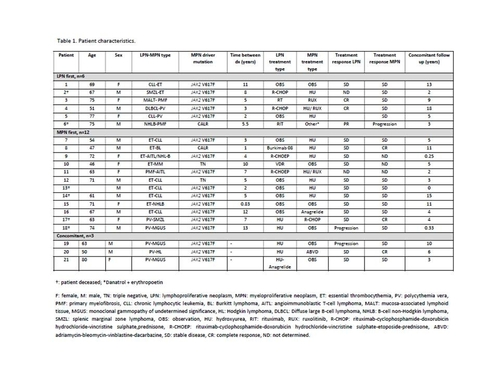
We report a series of 21 patients: 57% received an MPN dx first (average age at dx 66.4 years) then LPN, 28.5% received an LPN dx first (average age at dx 68.1 years) then MPN, while 14.5% had a concomitant dx (average age at dx 64.3 years). The most frequent LPN dx was chronic lymphocytic leukemia (CLL, 33%); the most frequent MPN dx was essential thrombocythemia (ET, 48%). Coexisting syndromes with the highest incidence were CLL-ET (28.6%) and PV-MGUS (14.3%). Patient characteristics are shown in Table 1.
Mutated MPN driver genes were JAK2 and CALR for 81% (n=3, all V617F) and 10% (n=2, 1 Type 1, 1 Type 2), respectively; 10% of patients were triple negative (n=2). The MPN driver mutation was always detected in the CD34+ population, although the allelic frequency (VAF) was slightly inferior compared to PB in 2 cases. In 1 TN patient no mutation was found in PB but the JAK2 mutation was detected in CD34+ cells (VAF 2.7%). In the CD19+ population, the MPN driver gene mutation was undetectable except for 1 patient who had a high VAF (CALR Type 1) in all cell populations and developed the LPN just one year after the initial MPN diagnosis. In the other 3 MPN-first cases, the LPN developed after 3-12 years.
With regard to other myeloid genes, TET2 was mutated in 2 patients in SP and CD34+ cells, while 1 patient was mutated in both DNMT3A and PTPN11. None of these variants were detected in the CD19+ populations. The only alteration observed in CD19+ cells was a chromosome 21 deletion (also present in the CD34+ population but not in PB).
Conclusion
In 1 patient the MPN driver mutation was present in the CD19+ population and the LPN debuted only one year after the MPN dx. In this case, the mutation was developed in an immature cell preceding myeloid/lymphoid differentiation and could have had a role in the development of LPN. In the rest of the patients, LPN development seemed to be independent of the JAK2/CALR driver gene mutation.
Our results show that the genetic analysis of CD34+ cells could help reveal the driver gene mutation in triple negative cases. The study of myeloid and lymphoid separated populations gives us a better understanding of the mechanisms of concomitant MPN/LPN development.
Keyword(s): Lymphoproliferative disorder, Myeloproliferative disorder
Abstract: EP1116
Type: E-Poster Presentation
Session title: Myeloproliferative neoplasms - Clinical
Background
The coexistence of myeloproliferative neoplasms (MPN) and lymphoproliferative neoplasms (LPN) is a rare finding. However, studies have demonstrated that patients with MPN have approximately a 3-fold higher risk of developing an LPN than the general population. The clonal origin of these diseases has yet to be elucidated, although there are suggestions that their origin may lie in multipotent hematopoietic stem cells.
Aims
Use of next-generation sequencing (NGS) to identify myeloid mutations in separated cellular populations associated with the development of coexisting MPN/LPN syndromes.
Methods
All patients with a MPN or LPN diagnosis (dx) between 1997 and 2020 were retrospectively analyzed for concomitant MPN/LPN according to the WHO 2016 criteria. Of the 21 patients identified, informed consent was obtained from the live patients (n=13) for peripheral blood (PB) extraction. CD34+ (progenitor cells), CD15+ (granulocytes), and CD19+ (B lymphocytes) cell populations were separated using an autoMACS Pro. So far, genomic DNA of 5 patients has been analyzed by NGS with the MiSeq (Illumina) platform using the targeted panel Myeloid Solution™ (SOPHiA GENETICS). Only variants with VAF ≥1%, MAF<1% and described as having a pathogenic or probably pathogenic effect were considered.
Results
We report a series of 21 patients: 57% received an MPN dx first (average age at dx 66.4 years) then LPN, 28.5% received an LPN dx first (average age at dx 68.1 years) then MPN, while 14.5% had a concomitant dx (average age at dx 64.3 years). The most frequent LPN dx was chronic lymphocytic leukemia (CLL, 33%); the most frequent MPN dx was essential thrombocythemia (ET, 48%). Coexisting syndromes with the highest incidence were CLL-ET (28.6%) and PV-MGUS (14.3%). Patient characteristics are shown in Table 1.
Mutated MPN driver genes were JAK2 and CALR for 81% (n=3, all V617F) and 10% (n=2, 1 Type 1, 1 Type 2), respectively; 10% of patients were triple negative (n=2). The MPN driver mutation was always detected in the CD34+ population, although the allelic frequency (VAF) was slightly inferior compared to PB in 2 cases. In 1 TN patient no mutation was found in PB but the JAK2 mutation was detected in CD34+ cells (VAF 2.7%). In the CD19+ population, the MPN driver gene mutation was undetectable except for 1 patient who had a high VAF (CALR Type 1) in all cell populations and developed the LPN just one year after the initial MPN diagnosis. In the other 3 MPN-first cases, the LPN developed after 3-12 years.
With regard to other myeloid genes, TET2 was mutated in 2 patients in SP and CD34+ cells, while 1 patient was mutated in both DNMT3A and PTPN11. None of these variants were detected in the CD19+ populations. The only alteration observed in CD19+ cells was a chromosome 21 deletion (also present in the CD34+ population but not in PB).
Conclusion
In 1 patient the MPN driver mutation was present in the CD19+ population and the LPN debuted only one year after the MPN dx. In this case, the mutation was developed in an immature cell preceding myeloid/lymphoid differentiation and could have had a role in the development of LPN. In the rest of the patients, LPN development seemed to be independent of the JAK2/CALR driver gene mutation.
Our results show that the genetic analysis of CD34+ cells could help reveal the driver gene mutation in triple negative cases. The study of myeloid and lymphoid separated populations gives us a better understanding of the mechanisms of concomitant MPN/LPN development.
Keyword(s): Lymphoproliferative disorder, Myeloproliferative disorder