

Contributions
Abstract: EP1108
Type: E-Poster Presentation
Session title: Myeloproliferative neoplasms - Clinical
Background
Absolute erythrocytosis is characterized by persistently raised Haemoglobin (Hb) and haematocrit (Ht) levels. Polycythaemia Vera (PV) is a clonal disease associated in 95% of cases with JAK2V617F mutation, while familial erythrocytoses are poorly understood.
Idiopathic erythrocytosis (IE) has not a definite cause, even after an accurate diagnostic investigation. Recently, also HFE mutations have been observed frequently in erythrocytotic patients. The recent availability of NGS technique has offered us the possibility to recognize mutations in known and unknown genes involved in the pathogenesis of erythrocytosis.
Aims
We report a venetian family with erythrocytosis in whom we found an undescribed association of mutations all consistent with the clinical phenotype.
Methods
The family comprehends a man, his wife, and their two children, a male and a female. Hematocrit, ferritin at diagnosis and necessity of phlebotomies were recorded if available. An NGS panel constituted by JAK 2, EGLN 1 (PHD2), HBB, HBA1, HBA2, ASXL1, HFE, HFE2, TFR2, HAMP, SLC40A1, SLC11A2, VHL, BPMG, EPAS1 was used.
Results
Results are showed in Table I.
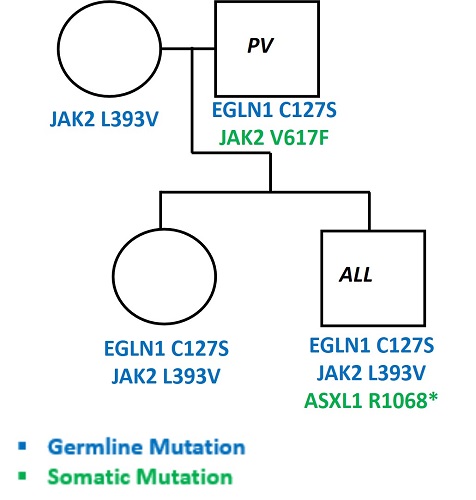
Genealogic tree of the family is reported in Figure I.
Member | Age | History | Somatic Mutations | Germline Mutations | HT (%) | Ferritin (µG/L) | Phlebotomies |
Father | 68 | Treated for PV since 2014, symptomatic for chest pain when Ht is high, no coronaropathy. | JAK2V617F | EGLN1 C127S | 56 | n.a. | 4/year since 2014, recently increasing |
Mother | 57 | Asymptomatic | - | JAK2 L393V | 40 | n.a. | no |
Son | 26 | Treated for acute lymphoblastic leukemia (ALL) in 2012 with high transfusion need and iron overload, developed erythrocytosis in 2017 | ASXL1 R1068* | EGLN1 C127S JAK2 L393V | 50 | 510 | 4/year since 2017 |
Daughter | 30 | Asymptomatic | - | EGLN1 C127S JAK2 L393V | n.a. | n.a. | no |
Conclusion
In this study we show the presence of contemporary mutations involved in erythrocytosis in more members of the same family.
Father and the two children have the same germline mutation (EGLN1 C127S) among with a mutation in JAK2 gene, somatic in the father (V617F) and germline in the two children (L393V). The mother has also this JAK2 germline mutation. The son has also a somatic mutation in ASXL1 R1068*. Interestingly, the son developed an acute lymphoblastic leukemia (ALL) at the age of 18 and developed erythrocytosis after recovery. The daughter and the mother are still asymptomatic.
It has been hypothesized that JAK2 germ line mutations may precede the acquisition of JAK2V617F lesion during the evolution of PV phenotype. The PHD2 mutation of this family is frequently observed in Tibetans, that establish an escape mechanism against erythrocytosis at high altitude, while in conditions of normoxia, the EGLN1 p.C127S variant has still an unclear function. We surmise that the association of PHD2 and JAK2 mutations could be associated with a more significant increase in hematocrit in this family. Moreover, usually ASXL1 is an epigenetic modifier, a marker of high risk for shorter survival in primary myelofibrosis (PMF). IE patients during their clinical history do not usually evolve in overt PV, nor in PMF or AML, and the role of ASXL1 mutation in these patients is therefore unknown.
In conclusion, the use of a specific NGS panel for erythrocytosis is a useful tool to explore new and combined genes variants and to study other members of the family to find the link between their haematological complications, in order to predict eventual clinical variations of the phenotype.
Keyword(s): Erythrocytosis, Familial
Abstract: EP1108
Type: E-Poster Presentation
Session title: Myeloproliferative neoplasms - Clinical
Background
Absolute erythrocytosis is characterized by persistently raised Haemoglobin (Hb) and haematocrit (Ht) levels. Polycythaemia Vera (PV) is a clonal disease associated in 95% of cases with JAK2V617F mutation, while familial erythrocytoses are poorly understood.
Idiopathic erythrocytosis (IE) has not a definite cause, even after an accurate diagnostic investigation. Recently, also HFE mutations have been observed frequently in erythrocytotic patients. The recent availability of NGS technique has offered us the possibility to recognize mutations in known and unknown genes involved in the pathogenesis of erythrocytosis.
Aims
We report a venetian family with erythrocytosis in whom we found an undescribed association of mutations all consistent with the clinical phenotype.
Methods
The family comprehends a man, his wife, and their two children, a male and a female. Hematocrit, ferritin at diagnosis and necessity of phlebotomies were recorded if available. An NGS panel constituted by JAK 2, EGLN 1 (PHD2), HBB, HBA1, HBA2, ASXL1, HFE, HFE2, TFR2, HAMP, SLC40A1, SLC11A2, VHL, BPMG, EPAS1 was used.
Results
Results are showed in Table I.
Genealogic tree of the family is reported in Figure I.
Member | Age | History | Somatic Mutations | Germline Mutations | HT (%) | Ferritin (µG/L) | Phlebotomies |
Father | 68 | Treated for PV since 2014, symptomatic for chest pain when Ht is high, no coronaropathy. | JAK2V617F | EGLN1 C127S | 56 | n.a. | 4/year since 2014, recently increasing |
Mother | 57 | Asymptomatic | - | JAK2 L393V | 40 | n.a. | no |
Son | 26 | Treated for acute lymphoblastic leukemia (ALL) in 2012 with high transfusion need and iron overload, developed erythrocytosis in 2017 | ASXL1 R1068* | EGLN1 C127S JAK2 L393V | 50 | 510 | 4/year since 2017 |
Daughter | 30 | Asymptomatic | - | EGLN1 C127S JAK2 L393V | n.a. | n.a. | no |
Conclusion
In this study we show the presence of contemporary mutations involved in erythrocytosis in more members of the same family.
Father and the two children have the same germline mutation (EGLN1 C127S) among with a mutation in JAK2 gene, somatic in the father (V617F) and germline in the two children (L393V). The mother has also this JAK2 germline mutation. The son has also a somatic mutation in ASXL1 R1068*. Interestingly, the son developed an acute lymphoblastic leukemia (ALL) at the age of 18 and developed erythrocytosis after recovery. The daughter and the mother are still asymptomatic.
It has been hypothesized that JAK2 germ line mutations may precede the acquisition of JAK2V617F lesion during the evolution of PV phenotype. The PHD2 mutation of this family is frequently observed in Tibetans, that establish an escape mechanism against erythrocytosis at high altitude, while in conditions of normoxia, the EGLN1 p.C127S variant has still an unclear function. We surmise that the association of PHD2 and JAK2 mutations could be associated with a more significant increase in hematocrit in this family. Moreover, usually ASXL1 is an epigenetic modifier, a marker of high risk for shorter survival in primary myelofibrosis (PMF). IE patients during their clinical history do not usually evolve in overt PV, nor in PMF or AML, and the role of ASXL1 mutation in these patients is therefore unknown.
In conclusion, the use of a specific NGS panel for erythrocytosis is a useful tool to explore new and combined genes variants and to study other members of the family to find the link between their haematological complications, in order to predict eventual clinical variations of the phenotype.
Keyword(s): Erythrocytosis, Familial