

Contributions
Abstract: EP1073
Type: E-Poster Presentation
Session title: Myeloproliferative neoplasms - Biology & Translational Research
Background
KRT-232 is a potent, selective, and orally bioavailable inhibitor of MDM2, a key negative regulator of the tumor suppressor protein, p53. KRT-232 restores p53 function resulting in apoptosis of malignant cells. Clinical trials investigating hematologic malignancies and solid tumors are ongoing. KRT-232 is a carboxylic acid that is metabolized primarily to an acyl glucuronide and undergoes enterohepatic recirculation. A 60 mg dose of KRT-232 given after a fast or a high-fat, high-calorie meal gave generally comparable KRT-232 pharmacokinetics (PK; Wong 2020). PBPK modeling was used to extrapolate this observation to higher doses and to define sensitivity of KRT-232 absorption to intrinsic and extrinsic factors.
Aims
1) Develop an absorption PBPK model for KRT-232; 2) Use the model to characterize regional absorption and fraction absorbed (Fa) across doses; 3) Predict whether PK exposure will be sensitive to gastric pH, gastrointestinal transit times, or meal type.
Methods
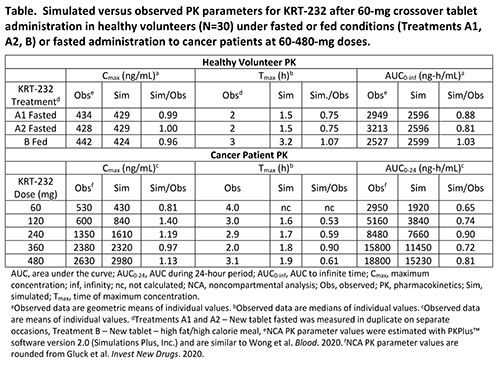
The PBPK model was developed in GastroPlus v9.7 (Simulations Plus, Inc.), using KRT-232 physicochemical properties that were mainly measured in vitro. Single dose healthy volunteer PK data (N=30) were from a 60-mg single-dose crossover study that compared a new tablet formulation administered after fasting and after a high-fat, high-calorie meal (Wong 2020). The model was verified against the single-dose clinical PK data, which led to a clearance adjustment. The final model was validated against PK results from a single ascending dose study in patients with advanced solid tumors or multiple myeloma (Gluck 2020, Ma 2019).
Results
Model simulations using the final model were consistent with observed data from healthy volunteers (60-mg single tablet dose) and from fasted cancer patients (60–480-mg single tablet dose; Table).
The model accurately reflected the lack of a meaningful change in KRT-232 PK after administration of a 60-mg tablet with a high-fat, high-calorie meal. KRT-232 Fa was near 100% and was not affected by dose or meal type. Absolute bioavailability was estimated to be ~73%, assuming UGT1A3 is the primary isoenzyme mediating KRT-232 metabolism. Under fasted conditions, most KRT-232 absorption occurred in the jejunum, with the relative amount of drug absorbed in the duodenum increasing slightly after a meal.
KRT-232 exposure was simulated for other meal types; Fa and AUC were not markedly affected. Over a physiologically-relevant range of values, sensitivity analyses showed that complete absorption of KRT-232 had little or no sensitivity to dose (≤ 480 mg), dissolution volume, stomach pH, or stomach transit time.
Simulated Fa of KRT-232 had little sensitivity to solubility from 1.6 μg/mL upwards past the reference solubility of the drug in Fasted and Fed State Simulated Intestinal Fluid (FaSSIF and FeSSIF, respectively [130 μg/mL]). Simulated fasted Fa was >90% with a mean particle radius almost 3-fold higher than that of the active pharmaceutical ingredient. Simulated Fa was even less sensitive to particle size under fed conditions.
Conclusion
The model adequately predicted KRT-232 PK under fasted and fed conditions. KRT-232 is completely absorbed across a wide range of doses, mainly from the jejunum. KRT-232 PK is not likely to be affected by proton pump inhibitor-mediated increases in gastric pH, in accord with its carboxylic acid structure. Because absorption is near 100% under most circumstances, major contributors to variability in KRT-232 PK will most likely be related to post-absorption intrinsic or extrinsic factors.
Keyword(s): Healthy individuals, P53, Pharmacokinetic
Abstract: EP1073
Type: E-Poster Presentation
Session title: Myeloproliferative neoplasms - Biology & Translational Research
Background
KRT-232 is a potent, selective, and orally bioavailable inhibitor of MDM2, a key negative regulator of the tumor suppressor protein, p53. KRT-232 restores p53 function resulting in apoptosis of malignant cells. Clinical trials investigating hematologic malignancies and solid tumors are ongoing. KRT-232 is a carboxylic acid that is metabolized primarily to an acyl glucuronide and undergoes enterohepatic recirculation. A 60 mg dose of KRT-232 given after a fast or a high-fat, high-calorie meal gave generally comparable KRT-232 pharmacokinetics (PK; Wong 2020). PBPK modeling was used to extrapolate this observation to higher doses and to define sensitivity of KRT-232 absorption to intrinsic and extrinsic factors.
Aims
1) Develop an absorption PBPK model for KRT-232; 2) Use the model to characterize regional absorption and fraction absorbed (Fa) across doses; 3) Predict whether PK exposure will be sensitive to gastric pH, gastrointestinal transit times, or meal type.
Methods
The PBPK model was developed in GastroPlus v9.7 (Simulations Plus, Inc.), using KRT-232 physicochemical properties that were mainly measured in vitro. Single dose healthy volunteer PK data (N=30) were from a 60-mg single-dose crossover study that compared a new tablet formulation administered after fasting and after a high-fat, high-calorie meal (Wong 2020). The model was verified against the single-dose clinical PK data, which led to a clearance adjustment. The final model was validated against PK results from a single ascending dose study in patients with advanced solid tumors or multiple myeloma (Gluck 2020, Ma 2019).
Results
Model simulations using the final model were consistent with observed data from healthy volunteers (60-mg single tablet dose) and from fasted cancer patients (60–480-mg single tablet dose; Table).
The model accurately reflected the lack of a meaningful change in KRT-232 PK after administration of a 60-mg tablet with a high-fat, high-calorie meal. KRT-232 Fa was near 100% and was not affected by dose or meal type. Absolute bioavailability was estimated to be ~73%, assuming UGT1A3 is the primary isoenzyme mediating KRT-232 metabolism. Under fasted conditions, most KRT-232 absorption occurred in the jejunum, with the relative amount of drug absorbed in the duodenum increasing slightly after a meal.
KRT-232 exposure was simulated for other meal types; Fa and AUC were not markedly affected. Over a physiologically-relevant range of values, sensitivity analyses showed that complete absorption of KRT-232 had little or no sensitivity to dose (≤ 480 mg), dissolution volume, stomach pH, or stomach transit time.
Simulated Fa of KRT-232 had little sensitivity to solubility from 1.6 μg/mL upwards past the reference solubility of the drug in Fasted and Fed State Simulated Intestinal Fluid (FaSSIF and FeSSIF, respectively [130 μg/mL]). Simulated fasted Fa was >90% with a mean particle radius almost 3-fold higher than that of the active pharmaceutical ingredient. Simulated Fa was even less sensitive to particle size under fed conditions.
Conclusion
The model adequately predicted KRT-232 PK under fasted and fed conditions. KRT-232 is completely absorbed across a wide range of doses, mainly from the jejunum. KRT-232 PK is not likely to be affected by proton pump inhibitor-mediated increases in gastric pH, in accord with its carboxylic acid structure. Because absorption is near 100% under most circumstances, major contributors to variability in KRT-232 PK will most likely be related to post-absorption intrinsic or extrinsic factors.
Keyword(s): Healthy individuals, P53, Pharmacokinetic