

Contributions
Abstract: EP1061
Type: E-Poster Presentation
Session title: Myeloproliferative neoplasms - Biology & Translational Research
Background
Primary Myelofibrosis (PMF) represents the paradigm of onco-inflammatory disorders. In fact, although its pathogenesis is cell-intrinsic and triggered by acquired somatic mutations in specific myeloid genes, cell-extrinsic effects exerted by the malignant clone via inflammatory mediators result in systemic complications (Jutzi & Mullally, 2020). Moreover, chronic inflammation elicits clonal evolution, in a self-perpetrating vicious cycle (Hasselbalch, Mediators Inflamm 2015).
CCL2 (also known as Monocyte Chemoattractant Protein-1) is one of the most potent immune-modulatory cytokines known to be elevated in PMF and exerts its biological effects by preferentially engaging its cognate receptor CCR2, activating a downstream signaling which includes G-proteins, MAPK/ERK, PI3K/Akt and JAK/STAT pathways (Lim, Oncotarget 2016).
SNPs in the regulatory regions of CCL2 gene account for the great inter-individual variability in CCL2 expression levels. Specifically, the rs1024611 (A>G) SNP of CCL2 leads to higher chemokine production upon inflammatory noxa (Tam, PlosOne 2012). We recently demonstrated that homozygosity for the G allele variant of this SNP is associated to reduced survival in PMF (Masselli, ASH 2019 abs#1689).
Aims
Here we sought to prove the functional relevance of this finding by assessing expression and function of CCL2/CCR2 axis in PMF and its down-regulation induced by immunomodulatory therapy with ruxolitinib.
Methods
MNCs from therapy naïve PMF stratified according to the CCL2 SNP (n. 25), and from PMF before (T0) and after Ruxolitinib start (T1→T3) (n. 6) were in part pelleted and in part cultured for 20h in 10% fetal bovine serum-enriched RPMI medium with 1.1 mg/ml of IL1-β as described (Rovin, BBRC 1999). Cells were then subjected to western blot for CCL2 expression. Immunomagnetically-isolated primary CD34+ cells from PMF and healthy donors (HD) were checked for CCR2 expression by flow cytometry and cultured for 24h in serum-free X-vivo medium with and without rhCCL2 (100 ng/mL). Cells were subjected to western blot to assess signaling pathways activation.
Results
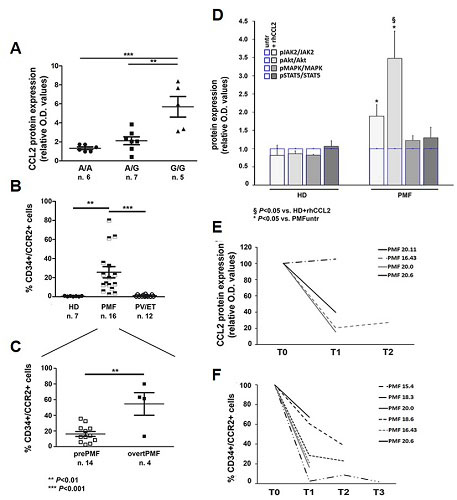
The rs1024611 SNP is functionally relevant in PMF since the G allele exerts a dose-dependent effect on CCL2 expression, with G/G PMF significantly overexpressing CCL2 as compared to the other genotypes (panel A). We asked whether PMF hematopoietic progenitors could be a target of CCL2 and we found that PMF CD34+ cells uniquely display CCR2 (> 60 fold-higher expression as compared to HD and >26 fold-higher as compared to PV and ET, which share with PMF the same driver mutations) (panel B). Additionally, among PMF, overtPMF significantly overexpress CCR2 as compared to prePMF (panel C). When PMF CD34+ cells were stimulated with rhCCL2, a significant phosphorylation of JAK2 and Akt was detected. As control, CCR2-negative CD34+ cells were subject to the same treatment and do not display CCL2-mediated phosphorylation of JAK/STAT, Akt and MAP-kinase pathways (panel D). Finally, we proved that ruxolitinib is effective in turning-off CCL2/CCR2 signaling. In fact, MNCs from patients at 1, 3 and 6 months of therapy (T1→T3) display a significantly reduced capacity to produce CCL2 upon inflammatory stimulus as compared to T0 (before ruxo start). A significant and stable reduction of the chemokine receptor on PMF CD34+ was detected as well (panel E-F).
Conclusion
CCL2/CCR2 chemokine axis is selectively activated in PMF, boosting pro-survival signals induced by driver mutations. Ruxolitinib effectively counteracts CCL2 production and down-regulates CCR2 expression in PMF cells.
Keyword(s): Chemokine, Myelofibrosis, Ruxolitinib, Signal transduction
Abstract: EP1061
Type: E-Poster Presentation
Session title: Myeloproliferative neoplasms - Biology & Translational Research
Background
Primary Myelofibrosis (PMF) represents the paradigm of onco-inflammatory disorders. In fact, although its pathogenesis is cell-intrinsic and triggered by acquired somatic mutations in specific myeloid genes, cell-extrinsic effects exerted by the malignant clone via inflammatory mediators result in systemic complications (Jutzi & Mullally, 2020). Moreover, chronic inflammation elicits clonal evolution, in a self-perpetrating vicious cycle (Hasselbalch, Mediators Inflamm 2015).
CCL2 (also known as Monocyte Chemoattractant Protein-1) is one of the most potent immune-modulatory cytokines known to be elevated in PMF and exerts its biological effects by preferentially engaging its cognate receptor CCR2, activating a downstream signaling which includes G-proteins, MAPK/ERK, PI3K/Akt and JAK/STAT pathways (Lim, Oncotarget 2016).
SNPs in the regulatory regions of CCL2 gene account for the great inter-individual variability in CCL2 expression levels. Specifically, the rs1024611 (A>G) SNP of CCL2 leads to higher chemokine production upon inflammatory noxa (Tam, PlosOne 2012). We recently demonstrated that homozygosity for the G allele variant of this SNP is associated to reduced survival in PMF (Masselli, ASH 2019 abs#1689).
Aims
Here we sought to prove the functional relevance of this finding by assessing expression and function of CCL2/CCR2 axis in PMF and its down-regulation induced by immunomodulatory therapy with ruxolitinib.
Methods
MNCs from therapy naïve PMF stratified according to the CCL2 SNP (n. 25), and from PMF before (T0) and after Ruxolitinib start (T1→T3) (n. 6) were in part pelleted and in part cultured for 20h in 10% fetal bovine serum-enriched RPMI medium with 1.1 mg/ml of IL1-β as described (Rovin, BBRC 1999). Cells were then subjected to western blot for CCL2 expression. Immunomagnetically-isolated primary CD34+ cells from PMF and healthy donors (HD) were checked for CCR2 expression by flow cytometry and cultured for 24h in serum-free X-vivo medium with and without rhCCL2 (100 ng/mL). Cells were subjected to western blot to assess signaling pathways activation.
Results
The rs1024611 SNP is functionally relevant in PMF since the G allele exerts a dose-dependent effect on CCL2 expression, with G/G PMF significantly overexpressing CCL2 as compared to the other genotypes (panel A). We asked whether PMF hematopoietic progenitors could be a target of CCL2 and we found that PMF CD34+ cells uniquely display CCR2 (> 60 fold-higher expression as compared to HD and >26 fold-higher as compared to PV and ET, which share with PMF the same driver mutations) (panel B). Additionally, among PMF, overtPMF significantly overexpress CCR2 as compared to prePMF (panel C). When PMF CD34+ cells were stimulated with rhCCL2, a significant phosphorylation of JAK2 and Akt was detected. As control, CCR2-negative CD34+ cells were subject to the same treatment and do not display CCL2-mediated phosphorylation of JAK/STAT, Akt and MAP-kinase pathways (panel D). Finally, we proved that ruxolitinib is effective in turning-off CCL2/CCR2 signaling. In fact, MNCs from patients at 1, 3 and 6 months of therapy (T1→T3) display a significantly reduced capacity to produce CCL2 upon inflammatory stimulus as compared to T0 (before ruxo start). A significant and stable reduction of the chemokine receptor on PMF CD34+ was detected as well (panel E-F).
Conclusion
CCL2/CCR2 chemokine axis is selectively activated in PMF, boosting pro-survival signals induced by driver mutations. Ruxolitinib effectively counteracts CCL2 production and down-regulates CCR2 expression in PMF cells.
Keyword(s): Chemokine, Myelofibrosis, Ruxolitinib, Signal transduction