

Contributions
Abstract: S267
Type: Oral Presentation
Session title: Changing the scene on thalassemias
Background
Thalassemias are characterized by ineffective erythropoiesis and hemolysis due to imbalanced production and precipitation of globin chains. Adenosine triphosphate (ATP) levels are reduced in thalassemic red blood cells (RBCs), despite increased energy demands to maintain RBC cell integrity. Mitapivat (AG-348) is an oral activator of RBC pyruvate kinase (PKR), an enzyme regulating ATP production via glycolysis. In mice, mitapivat increased ATP synthesis and survival of thalassemic RBCs. Mitapivat has been shown to increase hemoglobin (Hb) in patients (pts) with PK deficiency and sickle cell disease.
Aims
To report the results of the 24-week (wk) core period of a phase 2 open-label trial of mitapivat in adults with α- or β-non–transfusion-dependent thalassemia (NTDT; NCT03692052).
Methods
Key inclusion criteria: β-thalassemia ± α-globin gene mutations, Hb E β-thalassemia, or α-thalassemia (Hb H disease); Hb ≤10.0 g/dL; ≤5 RBC units transfused in prior 24 wks and none in 8 wks prior to study drug. Pts received mitapivat 50 mg orally twice daily (BID), then increased to 100 mg BID at Wk 6 visit, if tolerated. Primary endpoint was % pts who achieved a Hb response (increase ≥1.0 g/dL from baseline [BL] at any time between Wks 4–12, inclusive). Secondary endpoints included sustained Hb response (≥2 Hb increases of ≥1.0 g/dL vs BL during Wks 12–24 after achieving a primary Hb response), time to first ≥1.0 g/dL Hb increase, markers of hemolysis and erythropoiesis, and safety.
Results
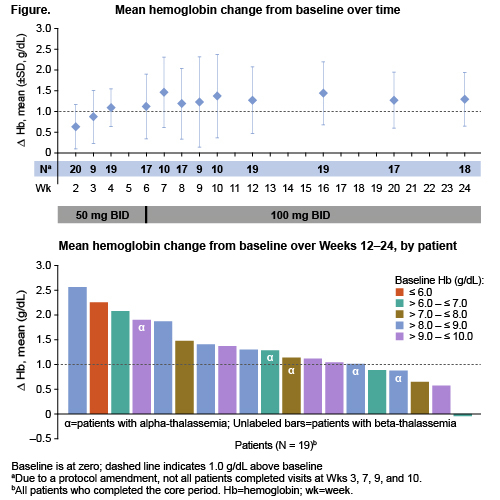
Of 20 pts (15 female) who received mitapivat, the mean age (SD) was 45.3 (11.8) yrs (range 29–67); mean (SD) BL Hb was 7.9 (1.4) g/dL (range 5.1–9.8). At Wk 6, all pts were escalated to 100 mg BID. Nineteen pts (95.0%) completed the core period, one discontinued due to adverse event (AE);17 pts continued to the extension period. Sixteen pts (80.0%, 90% CI 59.9, 92.9; p<0.0001) achieved a Hb response, including 5/5 pts with α-thalassemia and 11/15 pts with β-thalassemia. Mean (SD) time to first Hb increase >1.0 g/dL was 4.5 (3.2) wks, and mean Hb (SD) increase from BL during Wks 12–24 was 1.3 (0.6) g/dL. A sustained response was achieved in 65% of pts, including 5/5 pts with α-thalassemia and 8/15 pts with β-thalassemia. Mean Hb change from BL over time and average Hb increase from BL by patient are shown (Figure). Hb increases occurred across the spectrum of BL levels. Directional improvements in markers of erythropoiesis and hemolysis were also observed. The safety profile was consistent with that of previously published mitapivat studies. There was one serious grade 3 unrelated AE of renal impairment which led to treatment discontinuation. There were three non-serious grade 3 AEs leading to dose reduction: 1 resulted in a permanent dose reduction to 50 mg BID, and 2 were successfully rechallenged at 100 mg BID. The most common non-serious AEs occurring in ≥25% of pts were initial insomnia (10/20), dizziness (6/20), and headache (5/20). Dose escalation to 100 mg BID was well tolerated and not associated with any increase in AEs. Twelve pts had DXA scans at BL and 6 months; no notable trends for changes in bone mineral density were observed.
Conclusion
These results demonstrate that PKR activation with mitapivat was well tolerated and improved anemia, hemolysis, and ineffective erythropoiesis, and may represent a novel therapeutic approach for pts with α- or β-thalassemia. Pivotal trials will be initiated in 2021.
Keyword(s): AG-348, Hemolytic anemia, Pyruvate kinase, Thalassemia
Abstract: S267
Type: Oral Presentation
Session title: Changing the scene on thalassemias
Background
Thalassemias are characterized by ineffective erythropoiesis and hemolysis due to imbalanced production and precipitation of globin chains. Adenosine triphosphate (ATP) levels are reduced in thalassemic red blood cells (RBCs), despite increased energy demands to maintain RBC cell integrity. Mitapivat (AG-348) is an oral activator of RBC pyruvate kinase (PKR), an enzyme regulating ATP production via glycolysis. In mice, mitapivat increased ATP synthesis and survival of thalassemic RBCs. Mitapivat has been shown to increase hemoglobin (Hb) in patients (pts) with PK deficiency and sickle cell disease.
Aims
To report the results of the 24-week (wk) core period of a phase 2 open-label trial of mitapivat in adults with α- or β-non–transfusion-dependent thalassemia (NTDT; NCT03692052).
Methods
Key inclusion criteria: β-thalassemia ± α-globin gene mutations, Hb E β-thalassemia, or α-thalassemia (Hb H disease); Hb ≤10.0 g/dL; ≤5 RBC units transfused in prior 24 wks and none in 8 wks prior to study drug. Pts received mitapivat 50 mg orally twice daily (BID), then increased to 100 mg BID at Wk 6 visit, if tolerated. Primary endpoint was % pts who achieved a Hb response (increase ≥1.0 g/dL from baseline [BL] at any time between Wks 4–12, inclusive). Secondary endpoints included sustained Hb response (≥2 Hb increases of ≥1.0 g/dL vs BL during Wks 12–24 after achieving a primary Hb response), time to first ≥1.0 g/dL Hb increase, markers of hemolysis and erythropoiesis, and safety.
Results
Of 20 pts (15 female) who received mitapivat, the mean age (SD) was 45.3 (11.8) yrs (range 29–67); mean (SD) BL Hb was 7.9 (1.4) g/dL (range 5.1–9.8). At Wk 6, all pts were escalated to 100 mg BID. Nineteen pts (95.0%) completed the core period, one discontinued due to adverse event (AE);17 pts continued to the extension period. Sixteen pts (80.0%, 90% CI 59.9, 92.9; p<0.0001) achieved a Hb response, including 5/5 pts with α-thalassemia and 11/15 pts with β-thalassemia. Mean (SD) time to first Hb increase >1.0 g/dL was 4.5 (3.2) wks, and mean Hb (SD) increase from BL during Wks 12–24 was 1.3 (0.6) g/dL. A sustained response was achieved in 65% of pts, including 5/5 pts with α-thalassemia and 8/15 pts with β-thalassemia. Mean Hb change from BL over time and average Hb increase from BL by patient are shown (Figure). Hb increases occurred across the spectrum of BL levels. Directional improvements in markers of erythropoiesis and hemolysis were also observed. The safety profile was consistent with that of previously published mitapivat studies. There was one serious grade 3 unrelated AE of renal impairment which led to treatment discontinuation. There were three non-serious grade 3 AEs leading to dose reduction: 1 resulted in a permanent dose reduction to 50 mg BID, and 2 were successfully rechallenged at 100 mg BID. The most common non-serious AEs occurring in ≥25% of pts were initial insomnia (10/20), dizziness (6/20), and headache (5/20). Dose escalation to 100 mg BID was well tolerated and not associated with any increase in AEs. Twelve pts had DXA scans at BL and 6 months; no notable trends for changes in bone mineral density were observed.
Conclusion
These results demonstrate that PKR activation with mitapivat was well tolerated and improved anemia, hemolysis, and ineffective erythropoiesis, and may represent a novel therapeutic approach for pts with α- or β-thalassemia. Pivotal trials will be initiated in 2021.
Keyword(s): AG-348, Hemolytic anemia, Pyruvate kinase, Thalassemia