

Contributions
Abstract: S265
Type: Oral Presentation
Session title: Changing the scene on thalassemias
Background
β-Thalassemia is a common, frequently debilitating, inherited anemia caused by HBB gene mutations that reduce or eliminate the expression of the β-globin subunit of adult hemoglobin (HbA, α2β2). Consequently, excess free α-globin forms toxic precipitates during erythropoiesis, causing destruction of red blood cells (hemolysis) and apoptosis of their precursors (ineffective erythropoiesis [IE]). In many respects, β-thalassemia resembles other protein aggregation disorders in which the accumulation of unstable misfolded proteins leads to tissue destruction. Previously, we showed that free α-globin is eliminated by protein quality-control pathways, including the ubiquitin-proteasome system and autophagy (Khandros et al., Blood 2012). Recently, we identified that free α-globin is eliminated by ULK1-dependent, ATG5-independent autophagy that is suppressed by mTORC1 (Lechauve et al. Sci Transl Med 2019). We measured an elevated mTORC1 activity in β-thalassemic erythroid cells and the administration of the mTORC1 inhibitor rapamycin to β-thalassemic mice (strain HbbTh3/+) caused a reduction in α-globin precipitates with lessening of anemia and IE.
Aims
Our overall hypothesis is that inhibition of mTORC1 can alleviate β-thalassemia by stimulating ULK1-mediated autophagy of free α-globin. We will determine how disruption of the miR-144/451 locus impacts AMPK and mTORC1 activity in erythoird cells and how ablation of miR-144/451 mitigates β-thalassemia.
Methods
Our initial step has been to cross our mouse strains miR-144/451-/- and HbbTh3/+. miR-451, the most abundant erythroid microRNA, targets the mRNA encoding CAB39/MO25, a cofactor for the LKB1 kinase, which activates AMPK (Fang et al. Hematologica 2018). Moreover, activated-AMPK indirectly inhibits mTORC1 by phosphorylating TSC1/2. mTORC1 activity has been analyzed in maturation stage-matched erythroid precursors by immuno-flow cytometry and Western blotting. Afterwards we used mutliple readouts to assess β-thalassemia pathology including complete blood counts, electron microscopy of FACS-purified reticulocytes, RBC fractionation followed by gel electrophoresis to quantify α-globin precipitates, and assessment of extramedullary erythropoiesis in the spleen.
Results
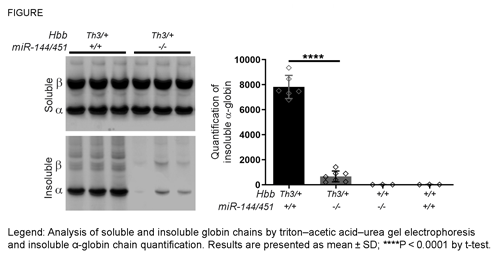
Here we show that genetic elimination of miR-451 alleviates β-thalassemia. Loss of miR-144/451 decreased erythroid mTORC1 activity in β-thalassemic erythroblasts by 33%, as measured by reduction of ribosomal protein S6 phosphorylation (P < 0.01). In β-thalassemic mice, elimination of miR-144/451 resulted in a 92% reduction of red blood cell α-globin precipitates (P < 0.0001; figure), with lessening of hemolysis and IE, as evidenced by 27% increased RBC count (P < 0.0001), 3-fold decreased reticulocyte count (P < 0.0001), and decreased spleen weight (0.52 ± 0.05 vs 0.22 ± 0.05 g, n=7; P < 0.0001) compared to β-thalassemic mice with intact miR-144/451 alleles. Importantly, the beneficial effects of miR-144/451 ablation were significantly reduced by genetic ablation of Ulk1 or Cab39, indicating that loss of miR-451 stimulates ULK1-mediated autophagy of free α-globin through the AMPK/mTORC1 pathway.
Conclusion
Advances in medical management and hematopoietic stem cell transplantation have improved and extended the lives of β-thalassemia patients significantly, however, new therapies are still required to further optimize care. Our findings suggest two novel therapeutic strategies to treat β-thalassemia by enhancing ULK1-mediated autophagy of free α-globin: pharmacological inhibition of mTORC1 or suppression of miR-451 by antagomirs.
Keyword(s): MTOR, Thalassemia
Abstract: S265
Type: Oral Presentation
Session title: Changing the scene on thalassemias
Background
β-Thalassemia is a common, frequently debilitating, inherited anemia caused by HBB gene mutations that reduce or eliminate the expression of the β-globin subunit of adult hemoglobin (HbA, α2β2). Consequently, excess free α-globin forms toxic precipitates during erythropoiesis, causing destruction of red blood cells (hemolysis) and apoptosis of their precursors (ineffective erythropoiesis [IE]). In many respects, β-thalassemia resembles other protein aggregation disorders in which the accumulation of unstable misfolded proteins leads to tissue destruction. Previously, we showed that free α-globin is eliminated by protein quality-control pathways, including the ubiquitin-proteasome system and autophagy (Khandros et al., Blood 2012). Recently, we identified that free α-globin is eliminated by ULK1-dependent, ATG5-independent autophagy that is suppressed by mTORC1 (Lechauve et al. Sci Transl Med 2019). We measured an elevated mTORC1 activity in β-thalassemic erythroid cells and the administration of the mTORC1 inhibitor rapamycin to β-thalassemic mice (strain HbbTh3/+) caused a reduction in α-globin precipitates with lessening of anemia and IE.
Aims
Our overall hypothesis is that inhibition of mTORC1 can alleviate β-thalassemia by stimulating ULK1-mediated autophagy of free α-globin. We will determine how disruption of the miR-144/451 locus impacts AMPK and mTORC1 activity in erythoird cells and how ablation of miR-144/451 mitigates β-thalassemia.
Methods
Our initial step has been to cross our mouse strains miR-144/451-/- and HbbTh3/+. miR-451, the most abundant erythroid microRNA, targets the mRNA encoding CAB39/MO25, a cofactor for the LKB1 kinase, which activates AMPK (Fang et al. Hematologica 2018). Moreover, activated-AMPK indirectly inhibits mTORC1 by phosphorylating TSC1/2. mTORC1 activity has been analyzed in maturation stage-matched erythroid precursors by immuno-flow cytometry and Western blotting. Afterwards we used mutliple readouts to assess β-thalassemia pathology including complete blood counts, electron microscopy of FACS-purified reticulocytes, RBC fractionation followed by gel electrophoresis to quantify α-globin precipitates, and assessment of extramedullary erythropoiesis in the spleen.
Results
Here we show that genetic elimination of miR-451 alleviates β-thalassemia. Loss of miR-144/451 decreased erythroid mTORC1 activity in β-thalassemic erythroblasts by 33%, as measured by reduction of ribosomal protein S6 phosphorylation (P < 0.01). In β-thalassemic mice, elimination of miR-144/451 resulted in a 92% reduction of red blood cell α-globin precipitates (P < 0.0001; figure), with lessening of hemolysis and IE, as evidenced by 27% increased RBC count (P < 0.0001), 3-fold decreased reticulocyte count (P < 0.0001), and decreased spleen weight (0.52 ± 0.05 vs 0.22 ± 0.05 g, n=7; P < 0.0001) compared to β-thalassemic mice with intact miR-144/451 alleles. Importantly, the beneficial effects of miR-144/451 ablation were significantly reduced by genetic ablation of Ulk1 or Cab39, indicating that loss of miR-451 stimulates ULK1-mediated autophagy of free α-globin through the AMPK/mTORC1 pathway.
Conclusion
Advances in medical management and hematopoietic stem cell transplantation have improved and extended the lives of β-thalassemia patients significantly, however, new therapies are still required to further optimize care. Our findings suggest two novel therapeutic strategies to treat β-thalassemia by enhancing ULK1-mediated autophagy of free α-globin: pharmacological inhibition of mTORC1 or suppression of miR-451 by antagomirs.
Keyword(s): MTOR, Thalassemia