

Contributions
Abstract: S261
Type: Oral Presentation
Session title: Changing the scene on sickle cell disease
Background
SCD is a progressive disease caused by a single point mutation in the β-globin gene. Despite lifelong treatment (tx) options, SCD results in painful vaso-occlusive events (VOEs), progressive vasculopathy, and chronic hemolytic anemia, leading to significant morbidity and early mortality. LentiGlobin for SCD (bb1111) gene therapy (GT) utilizes a modified human β-globin gene that produces GT-derived anti-sickling hemoglobin (Hb)AT87Q.
Aims
The ongoing Phase 1/2 HGB-206 study is the largest clinical trial of GT in SCD to date. Data from Group C are presented here.
Methods
Patients (pts; ≥12–≤50 yrs) with SCD and recurrent severe VOEs, including acute episodes of pain and acute chest syndrome, were enrolled. CD34+ cells were collected by plerixafor mobilization/apheresis and transduced with BB305 lentiviral vector. LentiGlobin was infused after myeloablative busulfan conditioning. Lab evaluations, SCD-related outcomes, pain intensity using Patient Reported Outcomes Measurement Information System (PROMIS)-57, and safety were assessed; data are median (min–max) unless otherwise stated.
Results
As of August 20, 2020, 43 pts (24 [12–38] yrs) had initiated cell collection, 32 of whom were treated with LentiGlobin and followed for 13.0 (1.1–30.9) months. Neutrophil and platelet engraftment were achieved at 19.5 (12–35) and 30 (18–136) days, respectively; all pts stopped RBC transfusions by 90 days post-tx. Near pancellular expression of HbAT87Q was observed ≥6 months post-tx with ~90% of RBCs containing βA-T87Q by 18 months (n=10). At last visit in evaluable pts with ≥6 months follow-up (n=22), total Hb was 12.0 (9.6–15.1) g/dL; HbAT87Q and HbS contribution to total Hb were 5.6 (2.7–8.9) and 6.1 (4.8–7.8) g/dL, respectively. At last visit in adolescents with ≥6 months follow-up (n=6), median total Hb and HbAT87Q were 13.5 and 6.1 g/dL, respectively.
At last visit, lactate dehydrogenase, indirect bilirubin, and reticulocytes (n=32) approached normalization along with reductions in nucleated RBCs and serum transferrin receptor levels (n=23).
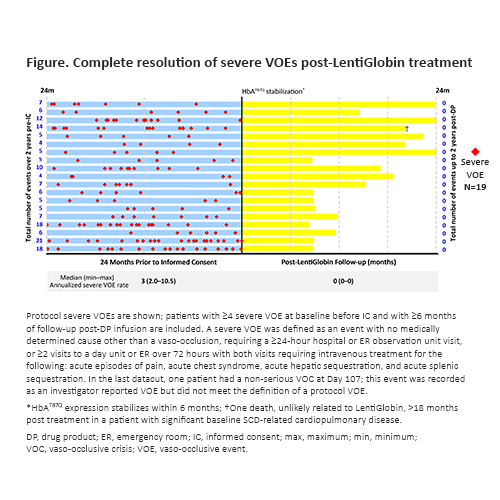
In 19 pts with a history of ≥4 severe VOEs and 6 months of follow-up, complete resolution of VOEs was observed after 6 months, with no reports of severe VOEs in the 24 months post-tx compared with an annualized severe VOE rate of 3 (2.0–10.5) in the 24 months prior to informed consent (Fig.). Overall mean PROMIS-57 pain intensity scores decreased from 4.5 at baseline (n=19) to 1.3 at Month 24 (n=4).
Tx-emergent serious adverse events (TESAEs) reported in 2 pts were abdominal pain, opioid withdrawal syndrome, nausea, and vomiting. All other TESAEs were reported in 1 pt. No SAEs were considered related to LentiGlobin. One pt had a nonserious Grade 2 event of febrile neutropenia considered to be LentiGlobin-related by the investigator. One death, unlikely related to LentiGlobin, occurred >18 months post-tx in a pt with significant baseline SCD-related cardiopulmonary disease. Beyond the August 20, 2020 data cutoff, one pt in Group C with persistent anemia 6 months after transplant was found to have trisomy 8 by fluorescence in situ hybridization on a 6-month bone marrow aspirate; the case is currently being investigated for myelodysplastic syndrome.
Conclusion
LentiGlobin tx resulted in complete resolution of severe VOEs up to 24 months post-tx and decreased patient-reported pain intensity from baseline. There was near pancellular HbAT87Q expression and improved SCD pathophysiology after tx. The safety profile of LentiGlobin tx regimen remains generally consistent with myeloablative single-agent busulfan conditioning and underlying SCD.
Keyword(s): Gene therapy, Lentiviral vector, Sickle cell disease
Abstract: S261
Type: Oral Presentation
Session title: Changing the scene on sickle cell disease
Background
SCD is a progressive disease caused by a single point mutation in the β-globin gene. Despite lifelong treatment (tx) options, SCD results in painful vaso-occlusive events (VOEs), progressive vasculopathy, and chronic hemolytic anemia, leading to significant morbidity and early mortality. LentiGlobin for SCD (bb1111) gene therapy (GT) utilizes a modified human β-globin gene that produces GT-derived anti-sickling hemoglobin (Hb)AT87Q.
Aims
The ongoing Phase 1/2 HGB-206 study is the largest clinical trial of GT in SCD to date. Data from Group C are presented here.
Methods
Patients (pts; ≥12–≤50 yrs) with SCD and recurrent severe VOEs, including acute episodes of pain and acute chest syndrome, were enrolled. CD34+ cells were collected by plerixafor mobilization/apheresis and transduced with BB305 lentiviral vector. LentiGlobin was infused after myeloablative busulfan conditioning. Lab evaluations, SCD-related outcomes, pain intensity using Patient Reported Outcomes Measurement Information System (PROMIS)-57, and safety were assessed; data are median (min–max) unless otherwise stated.
Results
As of August 20, 2020, 43 pts (24 [12–38] yrs) had initiated cell collection, 32 of whom were treated with LentiGlobin and followed for 13.0 (1.1–30.9) months. Neutrophil and platelet engraftment were achieved at 19.5 (12–35) and 30 (18–136) days, respectively; all pts stopped RBC transfusions by 90 days post-tx. Near pancellular expression of HbAT87Q was observed ≥6 months post-tx with ~90% of RBCs containing βA-T87Q by 18 months (n=10). At last visit in evaluable pts with ≥6 months follow-up (n=22), total Hb was 12.0 (9.6–15.1) g/dL; HbAT87Q and HbS contribution to total Hb were 5.6 (2.7–8.9) and 6.1 (4.8–7.8) g/dL, respectively. At last visit in adolescents with ≥6 months follow-up (n=6), median total Hb and HbAT87Q were 13.5 and 6.1 g/dL, respectively.
At last visit, lactate dehydrogenase, indirect bilirubin, and reticulocytes (n=32) approached normalization along with reductions in nucleated RBCs and serum transferrin receptor levels (n=23).
In 19 pts with a history of ≥4 severe VOEs and 6 months of follow-up, complete resolution of VOEs was observed after 6 months, with no reports of severe VOEs in the 24 months post-tx compared with an annualized severe VOE rate of 3 (2.0–10.5) in the 24 months prior to informed consent (Fig.). Overall mean PROMIS-57 pain intensity scores decreased from 4.5 at baseline (n=19) to 1.3 at Month 24 (n=4).
Tx-emergent serious adverse events (TESAEs) reported in 2 pts were abdominal pain, opioid withdrawal syndrome, nausea, and vomiting. All other TESAEs were reported in 1 pt. No SAEs were considered related to LentiGlobin. One pt had a nonserious Grade 2 event of febrile neutropenia considered to be LentiGlobin-related by the investigator. One death, unlikely related to LentiGlobin, occurred >18 months post-tx in a pt with significant baseline SCD-related cardiopulmonary disease. Beyond the August 20, 2020 data cutoff, one pt in Group C with persistent anemia 6 months after transplant was found to have trisomy 8 by fluorescence in situ hybridization on a 6-month bone marrow aspirate; the case is currently being investigated for myelodysplastic syndrome.
Conclusion
LentiGlobin tx resulted in complete resolution of severe VOEs up to 24 months post-tx and decreased patient-reported pain intensity from baseline. There was near pancellular HbAT87Q expression and improved SCD pathophysiology after tx. The safety profile of LentiGlobin tx regimen remains generally consistent with myeloablative single-agent busulfan conditioning and underlying SCD.
Keyword(s): Gene therapy, Lentiviral vector, Sickle cell disease