

Contributions
Abstract: S229
Type: Oral Presentation
Session title: Lymphoma - Translational research
Background
Heterogeneity in therapeutic response challenges the successful treatment of Diffuse Large B-Cell Lymphoma (DLBCL). Despite the recognition that DLBCL cases have diverse genetic and transcriptional characteristics, standard first-line therapy has remained unchanged for more than a decade. Further, cell-of-origin stratification into Germinal Centre (GC) and Activated B-cell (ABC) DLBCL fails to define much of the heterogeneity.
Canonical Nuclear Factor KappaB (NF-kB) is a dimeric transcription factor usually consisting of either cRel or RelA bound to p50. While aberrant NF-kB activation is frequently observed in DLBCL, subunit composition in individual DLBCL cases is not routinely characterized but has the potential to improve stratification and identify novel molecular targets for treatment. Computational simulations of NF-kB control over B-cell proliferation and apoptosis accurately predict experimental results with accuracy at both single-cell and cell-population scale. However, the key regulatory networks controlling B-cell differentiation were not factored into these predictive models.
Aims
1. Elucidate, with single-cell resolution, dynamic NF-kB control over B-cell fate decisions.
2. Develop a novel tool to enable rational therapeutic targeting of in B-cell lymphoma despite substantial heterogeneity.
Methods
Mechanistic systems biology approaches were combined with publically available data and targetted experiments designed to test computational predictions.
Results
Simulation of known regulatory interactions between NF-kB and the molecular networks controlling B-cell fate decisions revealed this molecular signalling network was insufficient to explain healthy B-cell differentiation. A computationally-driven prediction, that cRel must be downregulated during differentiation, lead to the discovery that although cRel drives B-cell proliferation it also blocks terminal differentiation to antibody-secreting plasma cells. We find that dynamic downregulation of cRel, through direct inhibition by Blimp1, is a pre-requisite for B-cell differentiation. The refined simulation, incorporating this novel interaction, accurately recapitulates B-cell population dynamics in wild-type (WT) and cRel knockout cells.
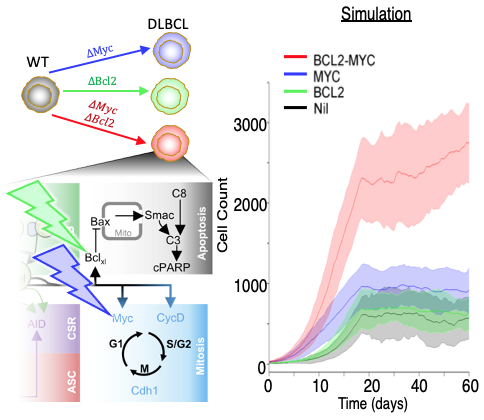
To establish whether a computational systems biology approach could enable accurate simulation of DLBCL we incorporated the aberrantly increased NF-kB activity seen in the disease. Interestingly, NF-kB sub-unit specific phenotypes emerge. Chronic cRel activity resulted in a differentiation block at the germinal centre stage, as seen in GC-DLBCL. RelA hyperactivity resulted in accumulation of a more differentiated cell type as see in ABC-DLBCL. To explore the potential of this approach to understand dysregulation beyond NF-kB, commonly-occurring mutations affecting MYC and BCL2 were simulated. While individual mutations only marginally perturbed cell counts the combination of MYC and BCL2 dysregulation commonly found in “double-hit” lymphoma had a striking synergistic effect in the model as seen in both experimental systems and clinical outcomes.
Conclusion
The ability of these computational systems biology models to accurately recapitulate clinically-observed emergent phenotypes from mutational changes enables the creation of mutation-specific “in silico” DLBCL laboratories. By incorporating the recently characterise vast mutational heterogeneity seen through whole exome sequencing DLBCL, we can use these models as tools to predict efficacious therapeutic approaches and biomarkers to stratify and treat lymphoma beyond current subtype classification.
Keyword(s): B cell, Diffuse large B cell lymphoma, NF- B
Abstract: S229
Type: Oral Presentation
Session title: Lymphoma - Translational research
Background
Heterogeneity in therapeutic response challenges the successful treatment of Diffuse Large B-Cell Lymphoma (DLBCL). Despite the recognition that DLBCL cases have diverse genetic and transcriptional characteristics, standard first-line therapy has remained unchanged for more than a decade. Further, cell-of-origin stratification into Germinal Centre (GC) and Activated B-cell (ABC) DLBCL fails to define much of the heterogeneity.
Canonical Nuclear Factor KappaB (NF-kB) is a dimeric transcription factor usually consisting of either cRel or RelA bound to p50. While aberrant NF-kB activation is frequently observed in DLBCL, subunit composition in individual DLBCL cases is not routinely characterized but has the potential to improve stratification and identify novel molecular targets for treatment. Computational simulations of NF-kB control over B-cell proliferation and apoptosis accurately predict experimental results with accuracy at both single-cell and cell-population scale. However, the key regulatory networks controlling B-cell differentiation were not factored into these predictive models.
Aims
1. Elucidate, with single-cell resolution, dynamic NF-kB control over B-cell fate decisions.
2. Develop a novel tool to enable rational therapeutic targeting of in B-cell lymphoma despite substantial heterogeneity.
Methods
Mechanistic systems biology approaches were combined with publically available data and targetted experiments designed to test computational predictions.
Results
Simulation of known regulatory interactions between NF-kB and the molecular networks controlling B-cell fate decisions revealed this molecular signalling network was insufficient to explain healthy B-cell differentiation. A computationally-driven prediction, that cRel must be downregulated during differentiation, lead to the discovery that although cRel drives B-cell proliferation it also blocks terminal differentiation to antibody-secreting plasma cells. We find that dynamic downregulation of cRel, through direct inhibition by Blimp1, is a pre-requisite for B-cell differentiation. The refined simulation, incorporating this novel interaction, accurately recapitulates B-cell population dynamics in wild-type (WT) and cRel knockout cells.
To establish whether a computational systems biology approach could enable accurate simulation of DLBCL we incorporated the aberrantly increased NF-kB activity seen in the disease. Interestingly, NF-kB sub-unit specific phenotypes emerge. Chronic cRel activity resulted in a differentiation block at the germinal centre stage, as seen in GC-DLBCL. RelA hyperactivity resulted in accumulation of a more differentiated cell type as see in ABC-DLBCL. To explore the potential of this approach to understand dysregulation beyond NF-kB, commonly-occurring mutations affecting MYC and BCL2 were simulated. While individual mutations only marginally perturbed cell counts the combination of MYC and BCL2 dysregulation commonly found in “double-hit” lymphoma had a striking synergistic effect in the model as seen in both experimental systems and clinical outcomes.
Conclusion
The ability of these computational systems biology models to accurately recapitulate clinically-observed emergent phenotypes from mutational changes enables the creation of mutation-specific “in silico” DLBCL laboratories. By incorporating the recently characterise vast mutational heterogeneity seen through whole exome sequencing DLBCL, we can use these models as tools to predict efficacious therapeutic approaches and biomarkers to stratify and treat lymphoma beyond current subtype classification.
Keyword(s): B cell, Diffuse large B cell lymphoma, NF- B