

Contributions
Abstract: PB1821
Type: Publication Only
Session title: Thalassemias
Background
Alpha thalassemia is characterized by the reduced or absence synthesis of alpha globin chains, most commonly due to delectional mutations. Hemoglobin Agrinio [α29(B10) Leu→Pro, CTG>CCG (α2)], is an infrequent nondelectional alfa thalasemia (α-Thal).
Aims
To describe an unusual case of neonatal anaemia and its diagnostical and therapeutical approach.
Methods
A systematic research of PubMed’s Medline was done, using de words Agrinio, neonatal and Thalassemia. Then, we analysed the case using electronic health records of our centre (previous parents informed consent), for history taking and laboratory data collection.
Results
Case description: A newborn girl was admitted to the neonatal intensive care unit because of respiratory distress showing severe microcytic anemia (haemoglobin 67 g/L, MCV 70.5 fl) and increased lactate dehydrogenase (6630 U/L, reference range 225-660 U/L). Rest of hemolysis parameters were normal. All the obstetric control had been passed with no incident, except for the finding of mild pericardial effusion and cardiomegaly in the third trimester ultrasound. Delivery proceed without incidence.
During her admission, moderate pulmonary hypertension (corrected at first 48 hours), and patent foramen ovale were found. Toxic, infectious diseases and RH/ABO incompatibility were ruled out. Peripheral blood smear only showed significant anisopoikilocytosis with the presence of microcytosis, hypochromic red blood cells, target cells, helmet red blood cells, and reticulocytes. Capillary electrophoresis of haemoglobin was normal.
At sixteen days of life, she was discharged with supportive oral iron. In the next two months, she needed to be admitted twice, due to hyporexia, rejection of feeds and general decline in general appearance, with severe anemia and moderate transfusion dependence.
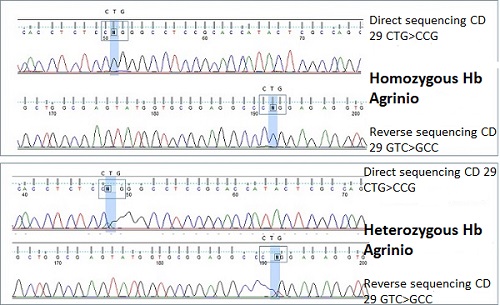
Given the consanguinity of the parents, both of gypsy ethnicity, and the suspicion of possible congenital haemolytic anaemia, thalassemia genetic study was requested to our reference centre, Hospital San Carlos, Madrid. After analysis, they concluded that the patient was a homozygous carrier of the CTG> CCG mutation in codon 29 of the first exon of the alpha 2 gene, which determines the change from leucine to proline at position 10 of the B helix of the alpha chain (haemoglobin Agrinio). Both parents were studied with diagnosis of heterozygous carriers of this mutation. None of her older siblings presented this mutation, so an HLA compatibility study has been carried out to consider the possibility of hematopoietic progenitors transplant in the future.
Conclusion
Discussion: Cases like the one presented, even today, pose a diagnostic and therapeutic challenge. Clinical suspicion (more in cases of the severity of ours), should guide us towards carrying out complementary tests quickly in order to be able to provide adequate treatment in time.
Agrinio haemoglobin is an unknown hemoglobinopathy, but molecular characterization of entities like this and the communication of results could help us make these patients increasingly less challenging in clinical practice.
Conclusion: In cases like the one presented, clinical suspicion is vital, and guided by this suspicion, genetic techniques and molecular diagnosis will be key to the definitive diagnostic orientation and the subsequent therapeutic approach.
Keyword(s): Anemia, Neonate, Thalassemia
Abstract: PB1821
Type: Publication Only
Session title: Thalassemias
Background
Alpha thalassemia is characterized by the reduced or absence synthesis of alpha globin chains, most commonly due to delectional mutations. Hemoglobin Agrinio [α29(B10) Leu→Pro, CTG>CCG (α2)], is an infrequent nondelectional alfa thalasemia (α-Thal).
Aims
To describe an unusual case of neonatal anaemia and its diagnostical and therapeutical approach.
Methods
A systematic research of PubMed’s Medline was done, using de words Agrinio, neonatal and Thalassemia. Then, we analysed the case using electronic health records of our centre (previous parents informed consent), for history taking and laboratory data collection.
Results
Case description: A newborn girl was admitted to the neonatal intensive care unit because of respiratory distress showing severe microcytic anemia (haemoglobin 67 g/L, MCV 70.5 fl) and increased lactate dehydrogenase (6630 U/L, reference range 225-660 U/L). Rest of hemolysis parameters were normal. All the obstetric control had been passed with no incident, except for the finding of mild pericardial effusion and cardiomegaly in the third trimester ultrasound. Delivery proceed without incidence.
During her admission, moderate pulmonary hypertension (corrected at first 48 hours), and patent foramen ovale were found. Toxic, infectious diseases and RH/ABO incompatibility were ruled out. Peripheral blood smear only showed significant anisopoikilocytosis with the presence of microcytosis, hypochromic red blood cells, target cells, helmet red blood cells, and reticulocytes. Capillary electrophoresis of haemoglobin was normal.
At sixteen days of life, she was discharged with supportive oral iron. In the next two months, she needed to be admitted twice, due to hyporexia, rejection of feeds and general decline in general appearance, with severe anemia and moderate transfusion dependence.
Given the consanguinity of the parents, both of gypsy ethnicity, and the suspicion of possible congenital haemolytic anaemia, thalassemia genetic study was requested to our reference centre, Hospital San Carlos, Madrid. After analysis, they concluded that the patient was a homozygous carrier of the CTG> CCG mutation in codon 29 of the first exon of the alpha 2 gene, which determines the change from leucine to proline at position 10 of the B helix of the alpha chain (haemoglobin Agrinio). Both parents were studied with diagnosis of heterozygous carriers of this mutation. None of her older siblings presented this mutation, so an HLA compatibility study has been carried out to consider the possibility of hematopoietic progenitors transplant in the future.
Conclusion
Discussion: Cases like the one presented, even today, pose a diagnostic and therapeutic challenge. Clinical suspicion (more in cases of the severity of ours), should guide us towards carrying out complementary tests quickly in order to be able to provide adequate treatment in time.
Agrinio haemoglobin is an unknown hemoglobinopathy, but molecular characterization of entities like this and the communication of results could help us make these patients increasingly less challenging in clinical practice.
Conclusion: In cases like the one presented, clinical suspicion is vital, and guided by this suspicion, genetic techniques and molecular diagnosis will be key to the definitive diagnostic orientation and the subsequent therapeutic approach.
Keyword(s): Anemia, Neonate, Thalassemia