

Contributions
Abstract: PB1767
Type: Publication Only
Session title: Sickle cell disease
Background
Sickle cell associated retinopathy is a well established chronic complication in sickle cell disease. There can be proliferative retinopathy (PR) and non-proliferative retinopathy (NPR). Although the risk of retinal changes is higher in heterozygotes SC (SC) disease followed by sickle-beta-thalassemia disease (S-tal) and homozygous sickle cell disease (SS), its exact incidence is unknown since most patients are asymptomatic.
Aims
Our study’s goals are, firstly to characterise sickle cell retinopathy in patients with SS, SC and S-tal disease and secondly understand if haematological parameters (such as hemoglobin level, haematocrit, mean corpuscular volume and percentage of haemoglobin S and F) can be used as a stratifying risk tool regarding presence of NPR and PR in SS patients.
Methods
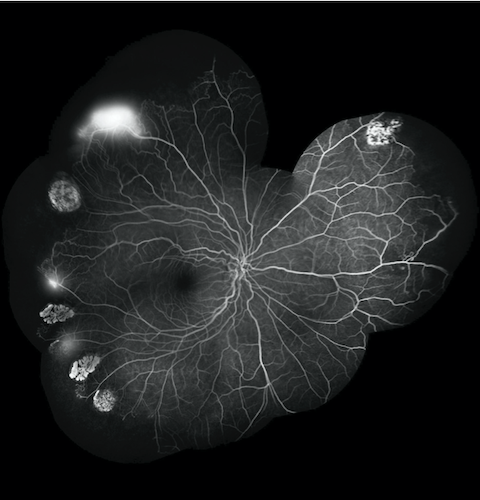
We included 106 patients followed between January/2017 and December/2019. Retinal evaluation was performed with binocular indirect ophthalmoscopy, fluorescein angiography (FA) and optical coherence tomography angiography (OCTA). In order to compare haematological parameters in the PR and NPR groups, we carried out a descriptive analysis of the variables and verification of normality. We resorted to the t-test of 2 independent samples for normal variables and the Mann-Whitney U test for variables without normal distribution. Subsequently, the existence of variables with an impact on the type of retinopathy was analysed through logistic regression
Results
We found 26 from 35 patients asymptomatic, however 13 (37.1%) had already fundoscopic changes, whilst 22 (62.9%) had no changes. Further ophthalmological evaluation with FA and OCTA, found that 29 patients (82.9%) had retinopathy. According to the Goldberg’s classification, 17 patients (58.6%) had NPR and 12 (41.4%) had PR. These patients included all with SC and S-tal disease (5 and 2, respectively) and 22 (78.6%) with SS disease.
Regarding the SS retinopathy group, these patients had higher levels of Hb S (69.2% vs. 67.21%) and lower levels of Hb F (9.54% vs. 13.47%). Statistical analyses revealed that the two populations PR and NPR, are similar, without significant statistical differences and with no specific associated parameters.
Conclusion
Our preliminary results reinforce that retinopathy is frequently clinically silent. Unfortunately, it is a well established under-diagnosed complication in sickle cell disease. Retinal complications should always be stratified with ophthalmological imaging techniques, since haematological parameters apparently are not a good stratifying risk tool. There is a minor difference regarding the amount of Hb S and F, but further work with a larger cohort of patient is needed. Therefore, our preliminary results reinforced that periodic ophthalmological evaluation is mandatory for all sickle cell disease patients and not only for the high-risk group sickle cell associated retinopathy.
Keyword(s): Complications, Hematocrit, Hemoglobin, Sickle cell anemia
Abstract: PB1767
Type: Publication Only
Session title: Sickle cell disease
Background
Sickle cell associated retinopathy is a well established chronic complication in sickle cell disease. There can be proliferative retinopathy (PR) and non-proliferative retinopathy (NPR). Although the risk of retinal changes is higher in heterozygotes SC (SC) disease followed by sickle-beta-thalassemia disease (S-tal) and homozygous sickle cell disease (SS), its exact incidence is unknown since most patients are asymptomatic.
Aims
Our study’s goals are, firstly to characterise sickle cell retinopathy in patients with SS, SC and S-tal disease and secondly understand if haematological parameters (such as hemoglobin level, haematocrit, mean corpuscular volume and percentage of haemoglobin S and F) can be used as a stratifying risk tool regarding presence of NPR and PR in SS patients.
Methods
We included 106 patients followed between January/2017 and December/2019. Retinal evaluation was performed with binocular indirect ophthalmoscopy, fluorescein angiography (FA) and optical coherence tomography angiography (OCTA). In order to compare haematological parameters in the PR and NPR groups, we carried out a descriptive analysis of the variables and verification of normality. We resorted to the t-test of 2 independent samples for normal variables and the Mann-Whitney U test for variables without normal distribution. Subsequently, the existence of variables with an impact on the type of retinopathy was analysed through logistic regression
Results
We found 26 from 35 patients asymptomatic, however 13 (37.1%) had already fundoscopic changes, whilst 22 (62.9%) had no changes. Further ophthalmological evaluation with FA and OCTA, found that 29 patients (82.9%) had retinopathy. According to the Goldberg’s classification, 17 patients (58.6%) had NPR and 12 (41.4%) had PR. These patients included all with SC and S-tal disease (5 and 2, respectively) and 22 (78.6%) with SS disease.
Regarding the SS retinopathy group, these patients had higher levels of Hb S (69.2% vs. 67.21%) and lower levels of Hb F (9.54% vs. 13.47%). Statistical analyses revealed that the two populations PR and NPR, are similar, without significant statistical differences and with no specific associated parameters.
Conclusion
Our preliminary results reinforce that retinopathy is frequently clinically silent. Unfortunately, it is a well established under-diagnosed complication in sickle cell disease. Retinal complications should always be stratified with ophthalmological imaging techniques, since haematological parameters apparently are not a good stratifying risk tool. There is a minor difference regarding the amount of Hb S and F, but further work with a larger cohort of patient is needed. Therefore, our preliminary results reinforced that periodic ophthalmological evaluation is mandatory for all sickle cell disease patients and not only for the high-risk group sickle cell associated retinopathy.
Keyword(s): Complications, Hematocrit, Hemoglobin, Sickle cell anemia