

Contributions
Abstract: PB1714
Type: Publication Only
Session title: Myeloproliferative neoplasms - Clinical
Background
Ruxolitinib (JAK1/JAK2 inhibitor) and fedratinib (JAK2/FLT3 inhibitor) are indicated in the United States for myelofibrosis (MF); however, some patients fail to achieve adequate/sustained response to initial JAK-inhibitor therapy. Itacitinib is a potent JAK inhibitor selective for JAK1 over JAK2 when administered as a once-daily sustained-release (SR) formulation. In a prior clinical trial, the SR formulation improved MF-related symptoms but had less effect with respect to spleen volume reduction (Mascarenhas JO, et al. Haematologica. 2017;102(2):327–335). When dosed as a twice-daily (BID) immediate release (IR) formulation, itacitinib, a selective JAK1 inhibitor, may attain sufficient JAK2 inhibition to better address the JAK2-mediated myeloproliferative features of the disease.
Aims
This open-label phase 2 study is designed to determine a tolerable and safe dose of itacitinib IR and whether that dose results in clinically significant reductions in symptoms and spleen volume in patients with MF who have previously received ruxolitinib and/or fedratinib monotherapy (INCB 39110-213; NCT04629508).
Methods
Eligible patients are aged ≥18 years, diagnosed with at least INT-1–risk (Dynamic International Prognostic Scoring System [Passamonti F, et al. Blood. 2010;115(9):1703–1708]) primary or secondary (post-polycythemia vera or post-essential thrombocythemia) MF and have received ruxolitinib and/or fedratinib monotherapy. Patients must also have palpable splenomegaly and platelets ≥50×109/L at screening. Exclusion criteria include receipt of a JAK inhibitor other than ruxolitinib or fedratinib, ≥10% myeloid blasts in peripheral blood or bone marrow, or inability to taper ruxolitinib/fedratinib over 14 days without use of other agents.
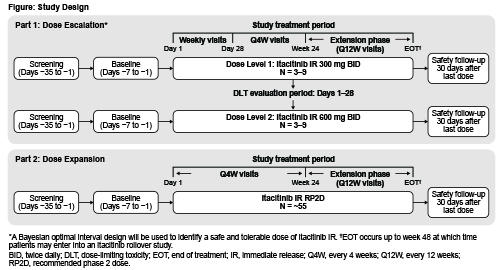
Two itacitinib IR dose levels (DLs) will be evaluated in part 1 following a Bayesian optimal interval design algorithm: 3–9 patients will be enrolled at DL1 (300 mg BID) and observed for 28 days for dose-limiting toxicity before enrollment at DL2 (600 mg BID) (Figure). Part 2 will enroll ~55 patients at the recommended phase 2 dose (RP2D) determined in part 1. Patients may remain on treatment until week 48 if they are receiving clinical benefit and have not met study withdrawal criteria. A safety follow-up will occur 30 days after treatment completion.
Primary objectives: part 1 - evaluate safety and tolerability of itacitinib IR and select the RP2D; part 2 - evaluate efficacy of itacitinib IR at the RP2D based on spleen volume reduction at week 24. Secondary objectives (part 2 only): evaluate safety and tolerability of itacitinib IR at the RP2D; evaluate efficacy of itacitinib IR at the RP2D with respect to MF symptom improvement at week 24 in patients with baseline total symptom score ≥10, quality-of-life improvement, and patient global impression of change. Sites are opening in the United States and Europe.
Results
Not applicable
Conclusion
Not applicable
Keyword(s): Janus Kinase inhibitor, Myelofibrosis, Myeloproliferative disorder
Abstract: PB1714
Type: Publication Only
Session title: Myeloproliferative neoplasms - Clinical
Background
Ruxolitinib (JAK1/JAK2 inhibitor) and fedratinib (JAK2/FLT3 inhibitor) are indicated in the United States for myelofibrosis (MF); however, some patients fail to achieve adequate/sustained response to initial JAK-inhibitor therapy. Itacitinib is a potent JAK inhibitor selective for JAK1 over JAK2 when administered as a once-daily sustained-release (SR) formulation. In a prior clinical trial, the SR formulation improved MF-related symptoms but had less effect with respect to spleen volume reduction (Mascarenhas JO, et al. Haematologica. 2017;102(2):327–335). When dosed as a twice-daily (BID) immediate release (IR) formulation, itacitinib, a selective JAK1 inhibitor, may attain sufficient JAK2 inhibition to better address the JAK2-mediated myeloproliferative features of the disease.
Aims
This open-label phase 2 study is designed to determine a tolerable and safe dose of itacitinib IR and whether that dose results in clinically significant reductions in symptoms and spleen volume in patients with MF who have previously received ruxolitinib and/or fedratinib monotherapy (INCB 39110-213; NCT04629508).
Methods
Eligible patients are aged ≥18 years, diagnosed with at least INT-1–risk (Dynamic International Prognostic Scoring System [Passamonti F, et al. Blood. 2010;115(9):1703–1708]) primary or secondary (post-polycythemia vera or post-essential thrombocythemia) MF and have received ruxolitinib and/or fedratinib monotherapy. Patients must also have palpable splenomegaly and platelets ≥50×109/L at screening. Exclusion criteria include receipt of a JAK inhibitor other than ruxolitinib or fedratinib, ≥10% myeloid blasts in peripheral blood or bone marrow, or inability to taper ruxolitinib/fedratinib over 14 days without use of other agents.
Two itacitinib IR dose levels (DLs) will be evaluated in part 1 following a Bayesian optimal interval design algorithm: 3–9 patients will be enrolled at DL1 (300 mg BID) and observed for 28 days for dose-limiting toxicity before enrollment at DL2 (600 mg BID) (Figure). Part 2 will enroll ~55 patients at the recommended phase 2 dose (RP2D) determined in part 1. Patients may remain on treatment until week 48 if they are receiving clinical benefit and have not met study withdrawal criteria. A safety follow-up will occur 30 days after treatment completion.
Primary objectives: part 1 - evaluate safety and tolerability of itacitinib IR and select the RP2D; part 2 - evaluate efficacy of itacitinib IR at the RP2D based on spleen volume reduction at week 24. Secondary objectives (part 2 only): evaluate safety and tolerability of itacitinib IR at the RP2D; evaluate efficacy of itacitinib IR at the RP2D with respect to MF symptom improvement at week 24 in patients with baseline total symptom score ≥10, quality-of-life improvement, and patient global impression of change. Sites are opening in the United States and Europe.
Results
Not applicable
Conclusion
Not applicable
Keyword(s): Janus Kinase inhibitor, Myelofibrosis, Myeloproliferative disorder