

Contributions
Abstract: PB1701
Type: Publication Only
Session title: Myeloproliferative neoplasms - Clinical
Background
Systemic Mastocytosis (SM) is a rare and heterogeneous disease characterized by the proliferation and accumulation of neoplastic mast cells in one or more organ systems. Its manifestations range from an indolent disease, to a highly aggressive neoplasm with associated multiorgan failure. Due to its rarity and wide range of symptoms, SM can be challenging to diagnose so a high index of suspicion should be maintained throughout the course of investigation of these patients.
Aims
To describe a single-center’s experience on the diagnosis and management of SM with special emphasis on its presenting manifestations and symptoms.
Methods
A retrospective revision of adult patients referred to our department and diagnosed with SM was performed. The diagnosis of SM followed the 2016 WHO classification criteria. Laboratory/diagnostic findings and patient management was carried out in collaboration with a specialized center.
Results
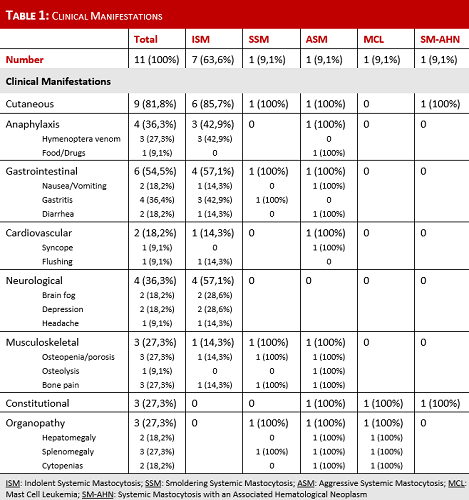
Eleven patients were diagnosed with SM at our institution, 5 females (45%) and 6 males (55%), with a median age at diagnosis of 52 years (range 38-68). Most patients (n=9, 82%) were referred to our clinic from other departments at our institution, while 2 patients (18%) were referred directly from their General Practitioner. Four patients (36%) presented with cutaneous manifestations, had a previous skin biopsy which confirmed mastocytosis in the skin, and were referred to exclude SM; 3 patients (27%) had previous severe anaphylaxis requiring hospitalization, 1 with cardiac arrest and admission to an Intensive Care Unit; 1 patient (9%) was referred for suspicion of an acute leukemia; the remainder 3 patients (27%) were referred for investigation of splenomegaly, polycythemia and eosinophilia. Regarding the diagnostic approach, all patients performed a complete blood count and basic biochemical evaluation, serum tryptase level measurement, bone marrow biopsy with morphologic and immunophenotypic study of mast cells. Nine patients (82%) had a KIT D816V mutation analysis. The presence of B and C findings was also evaluated. Hence 7 patients (64%) were diagnosed with Indolent SM (ISM) and 4 patients were diagnosed with other forms of SM, 1 case (9%) each: Smoldering SM (SSM); Aggressive SM (ASM); Mast Cell Leukemia (MCL); and SM with an associated hematological neoplasm (SM-AHN). The median time from first symptoms to diagnosis was 4 years, ranging from 3 months to 38 years. The main symptoms of our cohort are presented on Table 1. All patients received symptomatic treatment according to their needs, with H1 and H2 anti-histamines, sodium cromoglycate, biphosphonates, bee/wasp immunotherapy, omalizumab, among others. The patient with ASM received several cytoreductive therapies (pegylated interferon-α, cladribine, dasatinib), but only after initiation of midostaurin was the disease stabilized. The MCL patient was treated with cladribine with poor response, dying from refractory disease 1 month after diagnosis. All the remaining patients are still alive and under clinical surveillance.
Conclusion
Our data illustrates the heterogenicity of the clinical manifestations of SM. Patients range from nearly asymptomatic to severely ill, with a wide array of symptoms that can mimic other diseases. A multidisciplinary approach is of utmost importance for an earlier diagnosis of this rare condition and for a correct approach on the treatment options for these patients. Furthermore, an international collaborative work would certainly help further understand the clinical course of this disease and would be beneficial for the development of novel treatment strategies.
Keyword(s): Mast cell disease, Mastocytosis, Systemic mastocytosis
Abstract: PB1701
Type: Publication Only
Session title: Myeloproliferative neoplasms - Clinical
Background
Systemic Mastocytosis (SM) is a rare and heterogeneous disease characterized by the proliferation and accumulation of neoplastic mast cells in one or more organ systems. Its manifestations range from an indolent disease, to a highly aggressive neoplasm with associated multiorgan failure. Due to its rarity and wide range of symptoms, SM can be challenging to diagnose so a high index of suspicion should be maintained throughout the course of investigation of these patients.
Aims
To describe a single-center’s experience on the diagnosis and management of SM with special emphasis on its presenting manifestations and symptoms.
Methods
A retrospective revision of adult patients referred to our department and diagnosed with SM was performed. The diagnosis of SM followed the 2016 WHO classification criteria. Laboratory/diagnostic findings and patient management was carried out in collaboration with a specialized center.
Results
Eleven patients were diagnosed with SM at our institution, 5 females (45%) and 6 males (55%), with a median age at diagnosis of 52 years (range 38-68). Most patients (n=9, 82%) were referred to our clinic from other departments at our institution, while 2 patients (18%) were referred directly from their General Practitioner. Four patients (36%) presented with cutaneous manifestations, had a previous skin biopsy which confirmed mastocytosis in the skin, and were referred to exclude SM; 3 patients (27%) had previous severe anaphylaxis requiring hospitalization, 1 with cardiac arrest and admission to an Intensive Care Unit; 1 patient (9%) was referred for suspicion of an acute leukemia; the remainder 3 patients (27%) were referred for investigation of splenomegaly, polycythemia and eosinophilia. Regarding the diagnostic approach, all patients performed a complete blood count and basic biochemical evaluation, serum tryptase level measurement, bone marrow biopsy with morphologic and immunophenotypic study of mast cells. Nine patients (82%) had a KIT D816V mutation analysis. The presence of B and C findings was also evaluated. Hence 7 patients (64%) were diagnosed with Indolent SM (ISM) and 4 patients were diagnosed with other forms of SM, 1 case (9%) each: Smoldering SM (SSM); Aggressive SM (ASM); Mast Cell Leukemia (MCL); and SM with an associated hematological neoplasm (SM-AHN). The median time from first symptoms to diagnosis was 4 years, ranging from 3 months to 38 years. The main symptoms of our cohort are presented on Table 1. All patients received symptomatic treatment according to their needs, with H1 and H2 anti-histamines, sodium cromoglycate, biphosphonates, bee/wasp immunotherapy, omalizumab, among others. The patient with ASM received several cytoreductive therapies (pegylated interferon-α, cladribine, dasatinib), but only after initiation of midostaurin was the disease stabilized. The MCL patient was treated with cladribine with poor response, dying from refractory disease 1 month after diagnosis. All the remaining patients are still alive and under clinical surveillance.
Conclusion
Our data illustrates the heterogenicity of the clinical manifestations of SM. Patients range from nearly asymptomatic to severely ill, with a wide array of symptoms that can mimic other diseases. A multidisciplinary approach is of utmost importance for an earlier diagnosis of this rare condition and for a correct approach on the treatment options for these patients. Furthermore, an international collaborative work would certainly help further understand the clinical course of this disease and would be beneficial for the development of novel treatment strategies.
Keyword(s): Mast cell disease, Mastocytosis, Systemic mastocytosis