

Contributions
Abstract: PB1857
Type: Publication Only
Background
In CLL, the persistence of residual leukemic cells (MRD) after therapy is associated with poor outcome. Therefore, a better understanding of biological MRD characteristics would help to design newer and specific therapeutic approaches aimed at preventing clinical relapse. Nevertheless, working with a small amount of leukemic cells entails intensive labor to obtain enough material for gene expression profile studies. Here, we set forth an effective gene expression approach to study residual CLL cells.
Aims
The aim of this study was to set forth an effective gene expression approach to study residual CLL cells.
Methods
Leukocytes from fresh peripheral blood samples from 3 CLL patients were sorted based on MRD phenotype by multiparametric flow cytometry using a panel of 6 markers (CD43/CD5/CD19/CD81/CD79b/CD20). The quality and quantity of RNA isolated from two different inputs of cells 1x104 and 5 x105 were compared by using two silica columns protocols (RNeasy Micro and RNeasy Mini, Qiagen). Furthermore, RNA amplifications were carried out according to two manufacture protocols (Ovation Pico SL and Ovation Pico WTA system, Nugen) to further compare the quality and quantity of cDNA obtained. A total of 3.5µg of cDNA obtained with Pico WTA System was labeled with Encore Biotin Module (Nugen) and hybridized with GeneChip Hybridization Wash and Stain Kit (Thermo Fisher) to GeneChip Human Gene 2.0 ST arrays (Thermo Fisher Scientific). The reproducibility of arrays resulting from the different amount of input cells was compared.
Results
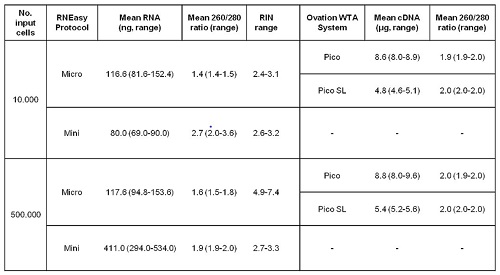
RNA extracted from 1x104 cells by RNeasy Micro and Mini Kit showed similar RNA Integrity Number (RIN), however, the amount of RNA obtained was higher using the RNeasy Micro Kit (mean: 116.0ng vs. 80.0ng). Of note, the quantity of RNA obtained from 1x104 cells was not significantly different from that isolated when the experiment was performed with a total of 5x105 cells.
Furthermore, RNA samples obtained after using RNeasy Micro Kit were amplified with two different kits. The amount of cDNA as a result of RNA amplification was enough to proceed for an array experiment by using any of the two different protocols (see table 1). In terms of cDNA quality, the mean 260/280 ratios were in all cases above 1.8. Nevertheless, samples processed by Ovation Pico SL WTA showed smaller amplified fragments that were not observed in samples processed by Ovation Pico WTA system. Microarray data analysis of RNA extracted from 1x104 and 5 x105 cells with Micro RNeasy and amplified with Ovation Pico WTA System, displayed a Pearson´s correlation average of 0.964.
Conclusion
By using the methodology herein presented, a total of 1x104 leukemic cells were enough to obtain quantity and quality samples to perform array studies. Thus, this methodology can be useful to carry out gene expression profiling experiments involving MRD in CLL.
Session topic: 5. Chronic lymphocytic leukemia and related disorders – Biology & Translational Research
Keyword(s): Arrays, Chronic Lymphocytic Leukemia, flow cytometry, Minimal residual disease (MRD)
Abstract: PB1857
Type: Publication Only
Background
In CLL, the persistence of residual leukemic cells (MRD) after therapy is associated with poor outcome. Therefore, a better understanding of biological MRD characteristics would help to design newer and specific therapeutic approaches aimed at preventing clinical relapse. Nevertheless, working with a small amount of leukemic cells entails intensive labor to obtain enough material for gene expression profile studies. Here, we set forth an effective gene expression approach to study residual CLL cells.
Aims
The aim of this study was to set forth an effective gene expression approach to study residual CLL cells.
Methods
Leukocytes from fresh peripheral blood samples from 3 CLL patients were sorted based on MRD phenotype by multiparametric flow cytometry using a panel of 6 markers (CD43/CD5/CD19/CD81/CD79b/CD20). The quality and quantity of RNA isolated from two different inputs of cells 1x104 and 5 x105 were compared by using two silica columns protocols (RNeasy Micro and RNeasy Mini, Qiagen). Furthermore, RNA amplifications were carried out according to two manufacture protocols (Ovation Pico SL and Ovation Pico WTA system, Nugen) to further compare the quality and quantity of cDNA obtained. A total of 3.5µg of cDNA obtained with Pico WTA System was labeled with Encore Biotin Module (Nugen) and hybridized with GeneChip Hybridization Wash and Stain Kit (Thermo Fisher) to GeneChip Human Gene 2.0 ST arrays (Thermo Fisher Scientific). The reproducibility of arrays resulting from the different amount of input cells was compared.
Results
RNA extracted from 1x104 cells by RNeasy Micro and Mini Kit showed similar RNA Integrity Number (RIN), however, the amount of RNA obtained was higher using the RNeasy Micro Kit (mean: 116.0ng vs. 80.0ng). Of note, the quantity of RNA obtained from 1x104 cells was not significantly different from that isolated when the experiment was performed with a total of 5x105 cells.
Furthermore, RNA samples obtained after using RNeasy Micro Kit were amplified with two different kits. The amount of cDNA as a result of RNA amplification was enough to proceed for an array experiment by using any of the two different protocols (see table 1). In terms of cDNA quality, the mean 260/280 ratios were in all cases above 1.8. Nevertheless, samples processed by Ovation Pico SL WTA showed smaller amplified fragments that were not observed in samples processed by Ovation Pico WTA system. Microarray data analysis of RNA extracted from 1x104 and 5 x105 cells with Micro RNeasy and amplified with Ovation Pico WTA System, displayed a Pearson´s correlation average of 0.964.
Conclusion
By using the methodology herein presented, a total of 1x104 leukemic cells were enough to obtain quantity and quality samples to perform array studies. Thus, this methodology can be useful to carry out gene expression profiling experiments involving MRD in CLL.
Session topic: 5. Chronic lymphocytic leukemia and related disorders – Biology & Translational Research
Keyword(s): Arrays, Chronic Lymphocytic Leukemia, flow cytometry, Minimal residual disease (MRD)