

Contributions
Abstract: PB1623
Type: Publication Only
Background
NOTCH/HES and AKT/mTOR pathways play important roles in the growth of T-lymphoblastic leukemia (T-ALL) cells. We have previously reported that treatment with γ-secretase inhibitor (GSI)-XXI, which prevents NOTCH activation, suppressed the growth of some T-ALL cell lines such as KOPT-K1, but promoted that of Jurkat cells, although both cell lines harbor activating NOTCH1 mutations. Given that Jurkat cells are PTEN-null, whereas KOPT-K1 cells are PTEN-wild type and that a NOTCH target HES1 downregulates PTEN expression, we hypothesized that the interplay among PTEN/AKT, NOTCH and its related pathways may regulate the growth of Jurkat cells.
Aims
To elucidate the mechanism underlying the induction of Jurkat cell growth by GSI, we examined the role of PTEN and its pseudogene PTENP1 reported to act as a tumor suppressor in some cancers.
Methods
Cells were transfected with small interfering RNAs targeting PTEN (siPTEN) and PTENP1 (siPTENP1), or control siRNA (siCont) by electroporation and analyzed for mRNA and protein expression by quantitative RT-PCR and immunoblotting, respectively. Cell growth was examined by a colorimetric WST-8 assay in three-day cultures; optical density (OD) was measured using an ELISA plate reader and cell growth was calculated as the percentage of the OD value normalized to that of siCont-transfected cells. To inhibit NOTCH activation, cells were treated with GSI-XXI.
Results
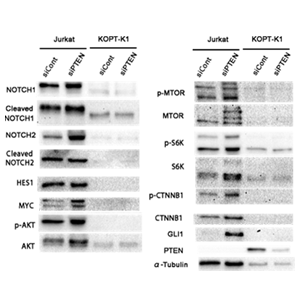
PTEN mRNA and protein were detected in KOPT-K1 cells but not in Jurkat cells. In KOPT-K1 cells, transfection with siPTEN inhibited PTEN mRNA and protein expression but did not affect cell growth; however, the proliferation of siPTEN-treated cells was slightly promoted by GSI treatment. In Jurkat cells, both siPTEN and siPTENP1 suppressed cell growth to 74% and 69% of control, respectively. Furthermore, transfection with siPTEN increased the expression of NOTCH, mTOR, WNT, and HEDGEHOG signaling proteins such as NOTCH1, cleaved NOTCH1, NOTCH2, MYC, AKT, p-AKT, p-mTOR, S6K, p-S6K, CTNNB1, p-CTNNB1, and GLI1 in Jurkat cells, but not in KOPT-K1 cells as shown in Figure.
Conclusion
Our study resulted in three interesting findings. First, siPTEN transfection suppressed the growth of PTEN-null Jurkat cells. One possible explanation is that siPTEN might bind to PTENP1 mRNA which has high sequence homology to PTEN mRNA; this notion is supported by the fact that transfection with siPTENP1 also suppressed Jurkat cell proliferation. Second, the inhibition of Jurkat cell growth by siPTEN was accompanied by the upregulation of cell proliferation-inducing proteins in NOTCH, mTOR, WNT, and HEDGEHOG pathways. This seeming discrepancy may be related to our previous finding that GSI treatment promoted the growth of Jurkat cells, i.e., that NOTCH activation apparently has suppressive effects on Jurkat cell proliferation. It is reported that PTEN expression is modulated by binding of some microRNAs and that PTENP1 transcript acts as a decoy for microRNA binding; therefore, siPTEN might downregulate PTENP1, which might affect the level of these growth-related microRNAs and cell growth. Third, GSI treatment promoted the proliferation of PTEN-knockdown KOPT-K1 cells similar to that of PTEN-null Jurkat cells, suggesting that cell growth stimulation by GSI may be associated with PTEN deficiency. Although our findings are not consistent with well-known mechanisms of NOTCH signaling, elucidation of these phenomena should further development of effective NOTCH-targeting therapies against T-ALL.
Session topic: 1. Acute lymphoblastic leukemia – Biology & Translational Research
Keyword(s): Notch, PTEN, SiRNA, T cell acute lymphoblastic leukemia
Abstract: PB1623
Type: Publication Only
Background
NOTCH/HES and AKT/mTOR pathways play important roles in the growth of T-lymphoblastic leukemia (T-ALL) cells. We have previously reported that treatment with γ-secretase inhibitor (GSI)-XXI, which prevents NOTCH activation, suppressed the growth of some T-ALL cell lines such as KOPT-K1, but promoted that of Jurkat cells, although both cell lines harbor activating NOTCH1 mutations. Given that Jurkat cells are PTEN-null, whereas KOPT-K1 cells are PTEN-wild type and that a NOTCH target HES1 downregulates PTEN expression, we hypothesized that the interplay among PTEN/AKT, NOTCH and its related pathways may regulate the growth of Jurkat cells.
Aims
To elucidate the mechanism underlying the induction of Jurkat cell growth by GSI, we examined the role of PTEN and its pseudogene PTENP1 reported to act as a tumor suppressor in some cancers.
Methods
Cells were transfected with small interfering RNAs targeting PTEN (siPTEN) and PTENP1 (siPTENP1), or control siRNA (siCont) by electroporation and analyzed for mRNA and protein expression by quantitative RT-PCR and immunoblotting, respectively. Cell growth was examined by a colorimetric WST-8 assay in three-day cultures; optical density (OD) was measured using an ELISA plate reader and cell growth was calculated as the percentage of the OD value normalized to that of siCont-transfected cells. To inhibit NOTCH activation, cells were treated with GSI-XXI.
Results
PTEN mRNA and protein were detected in KOPT-K1 cells but not in Jurkat cells. In KOPT-K1 cells, transfection with siPTEN inhibited PTEN mRNA and protein expression but did not affect cell growth; however, the proliferation of siPTEN-treated cells was slightly promoted by GSI treatment. In Jurkat cells, both siPTEN and siPTENP1 suppressed cell growth to 74% and 69% of control, respectively. Furthermore, transfection with siPTEN increased the expression of NOTCH, mTOR, WNT, and HEDGEHOG signaling proteins such as NOTCH1, cleaved NOTCH1, NOTCH2, MYC, AKT, p-AKT, p-mTOR, S6K, p-S6K, CTNNB1, p-CTNNB1, and GLI1 in Jurkat cells, but not in KOPT-K1 cells as shown in Figure.
Conclusion
Our study resulted in three interesting findings. First, siPTEN transfection suppressed the growth of PTEN-null Jurkat cells. One possible explanation is that siPTEN might bind to PTENP1 mRNA which has high sequence homology to PTEN mRNA; this notion is supported by the fact that transfection with siPTENP1 also suppressed Jurkat cell proliferation. Second, the inhibition of Jurkat cell growth by siPTEN was accompanied by the upregulation of cell proliferation-inducing proteins in NOTCH, mTOR, WNT, and HEDGEHOG pathways. This seeming discrepancy may be related to our previous finding that GSI treatment promoted the growth of Jurkat cells, i.e., that NOTCH activation apparently has suppressive effects on Jurkat cell proliferation. It is reported that PTEN expression is modulated by binding of some microRNAs and that PTENP1 transcript acts as a decoy for microRNA binding; therefore, siPTEN might downregulate PTENP1, which might affect the level of these growth-related microRNAs and cell growth. Third, GSI treatment promoted the proliferation of PTEN-knockdown KOPT-K1 cells similar to that of PTEN-null Jurkat cells, suggesting that cell growth stimulation by GSI may be associated with PTEN deficiency. Although our findings are not consistent with well-known mechanisms of NOTCH signaling, elucidation of these phenomena should further development of effective NOTCH-targeting therapies against T-ALL.
Session topic: 1. Acute lymphoblastic leukemia – Biology & Translational Research
Keyword(s): Notch, PTEN, SiRNA, T cell acute lymphoblastic leukemia