

Contributions
Abstract: PB1982
Type: Publication Only
Background
Hemoglobinopathies are the most frequent monogenetic alterations. The implantation of the “heel test” as a screening method in Spain has allowed the increase of new cases diagnostics. Hemoglobin (Hb) D most frequent variant is Hb D-Punjab. Hb D-Ouled Rabah, has been only studied as an anthropological entity with few studies of clinical significance. We present the case of two family members, the father in a homozygous state for Hb D and the son in a double heterozygous state (Hb D-Ouled Rabah/Hb S).
Aims
To describe the diagnosis process and clinical features of two Hb D- Ouled Rabah carrier patients, in homozygote state and Hb D-Ouled Rabah/Hb S double heterozygote state, in our hospital.
Methods
In March 2017, a newborn is referred to our outpatient clinic, with high suspicion of an abnormal hemoglobin (Hb) diagnostic in the heel-screening test (results were compatible with a B-Thalassemia/HbS in heterozygote state). A blood test confirmed the absence of acute hemolysis signs, with complete blood counts (CBC) and biochemistry tests in normal ranges. High-performance liquid chromatography (HPLC) was executed for the patient and both parents as the first study. Electrophoresis of hemoglobin in both, acid and alkaline media was also performed with no conclusive results. In order to achieve the diagnosis, molecular biology studies were necessary. The final reports were double heterozygote for the newborn (Hb D-Ouled Rabah [β19 Asn>Lys] and Hb S [β6 Glu>Val]) and Hb D-Ouled Rabah in homozygote state [β19 Asn>Lys] for the father.
Results
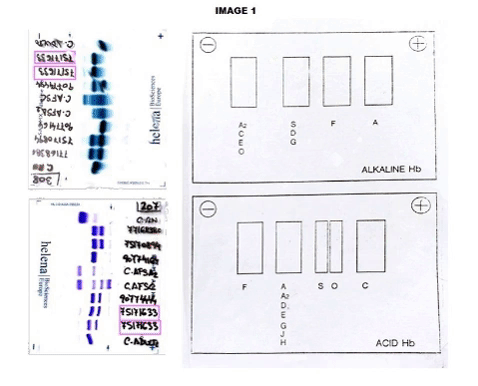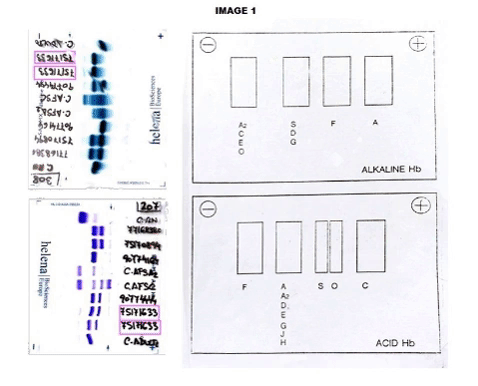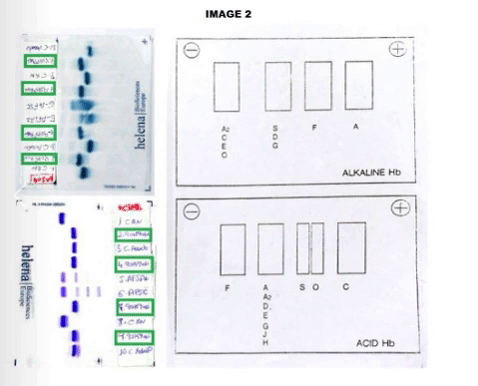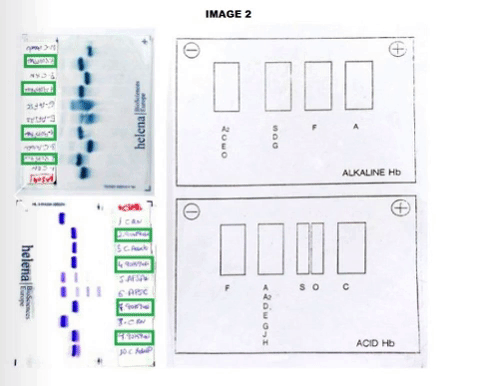
The CBC reveals Hb of 12.0 g/dL, hematocrit: 32.4%, mean corpuscular volume: 90.9 fL, reticulocyte count: 1.16%. Peripheral smear showed dyanocytes. The HPLC study in the patient showed Hb A2: 45%, Hb F: 5.7% and Hb S: 39%. Patient’s parents HPLC results: Father Hb A2: 77.3%, Hb F: 0.8%. Mother Result: Hb F: 0.8%, Hb A2: 3.6%, Hb S: 35%. In order to complete the study, an acidic and alkaline medium hemoglobin electrophoresis was accomplished. In the index case, we observed an Hb S in heterozygosis without being able to determine if he was also heterozygous for Hb G or Hb D. (Image 1) In the father case, we observed a band corresponding with a probable Hb G or Hb D in the homozygous state without being able to discern which of the two corresponding. (Image 2) The mother was carrying a Hemoglobin S in a heterozygous state.Given the inconclusive results, we conducted a molecular characterization of both, newborn and father abnormal hemoglobin. In the index case, it was described as a double heterozygous for a Hemoglobin D-Ouled Rabah [β19 Asn> Lys] and a Hemoglobin S [β6 Glu> Val], in the case of the progenitor, a Hemoglobin D-Ouled Rabah was found in homozygous state [β19 Asn> Lys]. Since there is no clinical information on the management of this type of cases, we decided to use the same prevention that we use in patients with a sickle cell disease.
Conclusion
The clinical behavior of Hb D-Punjab and Hb S in a double heterozygote state is well known but there is little knowledge about the hemolytic risk shared by Hb D-Ouled Rabah and Hb S when they occur together as in our patient. An adequate study of both electrophoresis and molecular biology in cases that are inconclusive will allow accurate diagnosis as well as an exhaustive description of the case. The monitoring of the evolution is of vital importance to predict the risk of hemolysis and acquire descriptive models to be able to use them as new cases appear.
Session topic: 29. Enzymopathies, membranopathies and other anemias
Keyword(s): Hemoglobin variants, Hemolysis, sickle cell disease
Abstract: PB1982
Type: Publication Only
Background
Hemoglobinopathies are the most frequent monogenetic alterations. The implantation of the “heel test” as a screening method in Spain has allowed the increase of new cases diagnostics. Hemoglobin (Hb) D most frequent variant is Hb D-Punjab. Hb D-Ouled Rabah, has been only studied as an anthropological entity with few studies of clinical significance. We present the case of two family members, the father in a homozygous state for Hb D and the son in a double heterozygous state (Hb D-Ouled Rabah/Hb S).
Aims
To describe the diagnosis process and clinical features of two Hb D- Ouled Rabah carrier patients, in homozygote state and Hb D-Ouled Rabah/Hb S double heterozygote state, in our hospital.
Methods
In March 2017, a newborn is referred to our outpatient clinic, with high suspicion of an abnormal hemoglobin (Hb) diagnostic in the heel-screening test (results were compatible with a B-Thalassemia/HbS in heterozygote state). A blood test confirmed the absence of acute hemolysis signs, with complete blood counts (CBC) and biochemistry tests in normal ranges. High-performance liquid chromatography (HPLC) was executed for the patient and both parents as the first study. Electrophoresis of hemoglobin in both, acid and alkaline media was also performed with no conclusive results. In order to achieve the diagnosis, molecular biology studies were necessary. The final reports were double heterozygote for the newborn (Hb D-Ouled Rabah [β19 Asn>Lys] and Hb S [β6 Glu>Val]) and Hb D-Ouled Rabah in homozygote state [β19 Asn>Lys] for the father.
Results
The CBC reveals Hb of 12.0 g/dL, hematocrit: 32.4%, mean corpuscular volume: 90.9 fL, reticulocyte count: 1.16%. Peripheral smear showed dyanocytes. The HPLC study in the patient showed Hb A2: 45%, Hb F: 5.7% and Hb S: 39%. Patient’s parents HPLC results: Father Hb A2: 77.3%, Hb F: 0.8%. Mother Result: Hb F: 0.8%, Hb A2: 3.6%, Hb S: 35%. In order to complete the study, an acidic and alkaline medium hemoglobin electrophoresis was accomplished. In the index case, we observed an Hb S in heterozygosis without being able to determine if he was also heterozygous for Hb G or Hb D. (Image 1) In the father case, we observed a band corresponding with a probable Hb G or Hb D in the homozygous state without being able to discern which of the two corresponding. (Image 2) The mother was carrying a Hemoglobin S in a heterozygous state.Given the inconclusive results, we conducted a molecular characterization of both, newborn and father abnormal hemoglobin. In the index case, it was described as a double heterozygous for a Hemoglobin D-Ouled Rabah [β19 Asn> Lys] and a Hemoglobin S [β6 Glu> Val], in the case of the progenitor, a Hemoglobin D-Ouled Rabah was found in homozygous state [β19 Asn> Lys]. Since there is no clinical information on the management of this type of cases, we decided to use the same prevention that we use in patients with a sickle cell disease.
Conclusion
The clinical behavior of Hb D-Punjab and Hb S in a double heterozygote state is well known but there is little knowledge about the hemolytic risk shared by Hb D-Ouled Rabah and Hb S when they occur together as in our patient. An adequate study of both electrophoresis and molecular biology in cases that are inconclusive will allow accurate diagnosis as well as an exhaustive description of the case. The monitoring of the evolution is of vital importance to predict the risk of hemolysis and acquire descriptive models to be able to use them as new cases appear.
Session topic: 29. Enzymopathies, membranopathies and other anemias
Keyword(s): Hemoglobin variants, Hemolysis, sickle cell disease