

Contributions
Abstract: S1591
Type: Oral Presentation
Presentation during EHA23: On Sunday, June 17, 2018 from 08:15 - 08:30
Location: Room A10
Background
The tumor suppressor gene p53 is pivotal for DNA damage response pathways by forcing cell cycle arrest, DNA repair and apoptosis. Hence, p53 is among the most frequently altered genes in cancer and its mutations are associated with large numbers of hematopoietic neoplasms, including MDS and AML. In hematopoietic stem cells (HSCs) p53 has been reported to be crucial for self-renewal, senescence and quiescence. Considering that very often leukemia emerges from stem and progenitor cells, it is remarkable that it still remains unclear how p53 activity prevents their transition to leukemia.
Aims
To investigate mechanisms that facilitate genomic and functional stability of the HSC pool and how p53 deficiency alters the functionality and the mutational landscape of HSCs in response to DNA damage.
Methods
We employed P53-/- mice together with wild type C57BL/6 mice to examine in vivo changes in cell cycle distribution and apoptosis levels of HSCs and less primitive progenitors in response to DNA damage. In this context, we also applied several assays to assess DNA repair pathways and changes in functionality of these cells. We visualized single HSCs within their bones by whole-mount immunofluorescence staining following confocal imaging. We performed transplantation experiments to study the consequences of HSC-associated DNA damage on the recipient mice. The detection of single point mutations and insertions/deletions was performed by sorting donor-derived HSCs following RNA-seq analysis and a bioinformatic approach which extracts information about mutation frequencies out of these data.
Results
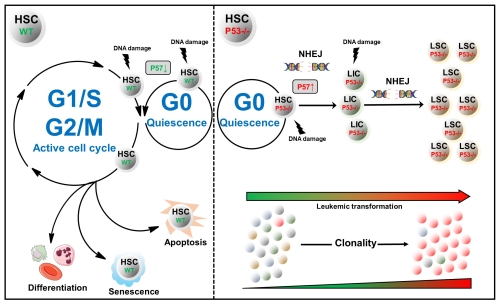
Here, we report a surprising new role of p53 in ensuring the integrity of HSCs: As previously reported, upon DNA damage HSCs from wild type mice enter the active cell cycle while simultaneously forcing apoptosis. HSCs from mice devoid of p53, however, preserve their quiescent state, protecting them from cell death. These HSCs exhibit a high abundance of 53BP1 foci suggesting a constitutive preference for the error prone non-homologues end joining pathway. These features are of cell-intrinsic and niche-independent nature, primarily stem cell specific and not present in more committed progenitors irrespective of p53. We demonstrate that p53-/- HSCs display persistently high P57(Kip2) levels after DNA damage offering a functional explanation for the observed phenotype. Indeed, P53/P57 double deficient HSCs react similar to irradiation induced DNA damage in comparison to their wild type counterparts. Importantly, transplanted p53-/- HSCs engraft massively in the bone marrow several months after sublethal irradiation, severely lose their differentiation potential and the corresponding recipient mice develop early symptoms of leukemia, such as increased spleen sizes and high numbers of blast cells in the bone marrow. Strikingly, the single point mutation frequency of P53-deficient HSCs isolated from these mice is not notably high whereas we observe a global increase of insertions/deletions near splice sites and intron regions of protein coding genes. These observations imply that initial steps of transformation are not solely driven by the accumulation of mutations but specific changes in the mutational landscape stimulate the transformation process of HSCs.
Conclusion
In compliance with our previously published data we believe that we have found evidence for a novel p53-dependent pathway, forcing DNA-damaged HSCs out of quiescence to enable their removal. This proposed mechanism may turn out to play a crucial role in preventing the leukemic transformation of these cells.
Session topic: 24. Hematopoiesis, stem cells and microenvironment
Keyword(s): Acute Myeloid Leukemia, DNA Damage, HSC, mutation analysis
Abstract: S1591
Type: Oral Presentation
Presentation during EHA23: On Sunday, June 17, 2018 from 08:15 - 08:30
Location: Room A10
Background
The tumor suppressor gene p53 is pivotal for DNA damage response pathways by forcing cell cycle arrest, DNA repair and apoptosis. Hence, p53 is among the most frequently altered genes in cancer and its mutations are associated with large numbers of hematopoietic neoplasms, including MDS and AML. In hematopoietic stem cells (HSCs) p53 has been reported to be crucial for self-renewal, senescence and quiescence. Considering that very often leukemia emerges from stem and progenitor cells, it is remarkable that it still remains unclear how p53 activity prevents their transition to leukemia.
Aims
To investigate mechanisms that facilitate genomic and functional stability of the HSC pool and how p53 deficiency alters the functionality and the mutational landscape of HSCs in response to DNA damage.
Methods
We employed P53-/- mice together with wild type C57BL/6 mice to examine in vivo changes in cell cycle distribution and apoptosis levels of HSCs and less primitive progenitors in response to DNA damage. In this context, we also applied several assays to assess DNA repair pathways and changes in functionality of these cells. We visualized single HSCs within their bones by whole-mount immunofluorescence staining following confocal imaging. We performed transplantation experiments to study the consequences of HSC-associated DNA damage on the recipient mice. The detection of single point mutations and insertions/deletions was performed by sorting donor-derived HSCs following RNA-seq analysis and a bioinformatic approach which extracts information about mutation frequencies out of these data.
Results
Here, we report a surprising new role of p53 in ensuring the integrity of HSCs: As previously reported, upon DNA damage HSCs from wild type mice enter the active cell cycle while simultaneously forcing apoptosis. HSCs from mice devoid of p53, however, preserve their quiescent state, protecting them from cell death. These HSCs exhibit a high abundance of 53BP1 foci suggesting a constitutive preference for the error prone non-homologues end joining pathway. These features are of cell-intrinsic and niche-independent nature, primarily stem cell specific and not present in more committed progenitors irrespective of p53. We demonstrate that p53-/- HSCs display persistently high P57(Kip2) levels after DNA damage offering a functional explanation for the observed phenotype. Indeed, P53/P57 double deficient HSCs react similar to irradiation induced DNA damage in comparison to their wild type counterparts. Importantly, transplanted p53-/- HSCs engraft massively in the bone marrow several months after sublethal irradiation, severely lose their differentiation potential and the corresponding recipient mice develop early symptoms of leukemia, such as increased spleen sizes and high numbers of blast cells in the bone marrow. Strikingly, the single point mutation frequency of P53-deficient HSCs isolated from these mice is not notably high whereas we observe a global increase of insertions/deletions near splice sites and intron regions of protein coding genes. These observations imply that initial steps of transformation are not solely driven by the accumulation of mutations but specific changes in the mutational landscape stimulate the transformation process of HSCs.
Conclusion
In compliance with our previously published data we believe that we have found evidence for a novel p53-dependent pathway, forcing DNA-damaged HSCs out of quiescence to enable their removal. This proposed mechanism may turn out to play a crucial role in preventing the leukemic transformation of these cells.
Session topic: 24. Hematopoiesis, stem cells and microenvironment
Keyword(s): Acute Myeloid Leukemia, DNA Damage, HSC, mutation analysis