

Contributions
Abstract: S881
Type: Oral Presentation
Presentation during EHA23: On Saturday, June 16, 2018 from 16:00 - 16:15
Location: Room A8
Background
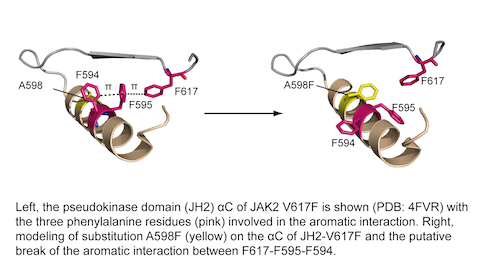
Dysfunction of JAK2 signaling is associated with hematopoietic malignancies. The JAK2 V617F mutation, which constitutively activates JAK2, is found in over 95% of Polycythemia Vera patients. JAK2 has a pivotal physiologic role in triggering several signaling pathways by binding to several cytokine receptors; therefore specific targeting of the mutant protein is absolutely required. Currently, no drug can discriminate between the mutant and the normal protein in myeloproliferative neoplasms. We have previously shown that an aromatic interaction between F617 and F594/F595 of pseudokinase domain helix C is required for constitutive signaling of JAK2 V617F, which constitutes the first molecular step in triggering hyperactivation in JAK2.
Aims
We aimed to find novel inhibitory strategies that directly destabilize the initial molecular step in V617F-mediated activation, namely the aromatic stacking between F617 and F594/F595 that only exist in the mutant conformation. We also aim to assess these approaches on the homologous JAK1 V658F, associated with ALL, and in the context of distinct cytokine receptors.
Methods
Via structure-guided mutagenesis, we assessed the role of key residues around the F617 aromatic interaction and used a combination of transcriptional activity luciferase-based assays, biochemical assays and cellular growth to measure the activity of JAKs in reconstituted JAK2 and JAK1 deficient cells.
Results
We created several aromatic mutations (S591F, A597F, A598F, S599F, V615F) strategically localizing around this aromatic interaction. We show that the specific mutation A598F preferentially inhibits JAK2 V617F over JAK2 WT, and without altering the normal function of the protein. Of importance, the inhibitory mutation A598F suppressed the hyperactivity of JAK2 V617F and other disease-associated JAK2 mutants such as JAK2 T875N and R683G, but not the JAK2 exon 12 mutant K539L.
We found that the mutation A598F and the homologous A639F can inhibit basal JAK2 V617F and JAK1 V658F, respectively, in the context of distinct cytokine receptors, such as IL2-R, IL9-R, IFNGR and type I IFN receptor (IFNAR).
Surprisingly, we found that the A598F mutation, as well as several other inhibitory mutations for JAK2 V617F, when in the background of the wild-type protein, drastically altered the cytokine responsiveness of the IFNGR, while it preserved normal cytokine response for EpoR, IL2-R, IL9R and IFNAR. This was even more compelling for JAK1 signaling with IFNGR; when these homologous inhibitory mutations were present in JAK1, they completely blocked the IFNγ signal transduction. Thus, it appears that the region of the JH2 helix C, which is required for JAK2 V617F and JAK1 V658F hyperactivation mechanism, is also crucial in specifically relaying the activation by cytokine in the heterodimeric IFNGR complex. Finally, we also show an unexpectedly low effect of JAK2 V617F on IFNGR complex compared to other cytokine receptors that physiologically utilize JAK2.
Conclusion
We identified a novel strategy to specifically inhibit JAK2 V617F and homologous JAK1 mutants, and we described an unexpected requirement for pseudokinase domain helix C specifically in the function of IFNGR. Inhibitory mutations localizing in the vicinity of JH2 helix aromatic interaction of JAK2 V617F restore normal function for most of JAK2-utilizing cytokine receptors except for the IFNGR.
Session topic: 15. Myeloproliferative neoplasms – Biology & Translational Research
Keyword(s): IFN-gamma, Janus Kinase inhibitor, Myeloproliferative disorder, Signal transduction
Abstract: S881
Type: Oral Presentation
Presentation during EHA23: On Saturday, June 16, 2018 from 16:00 - 16:15
Location: Room A8
Background
Dysfunction of JAK2 signaling is associated with hematopoietic malignancies. The JAK2 V617F mutation, which constitutively activates JAK2, is found in over 95% of Polycythemia Vera patients. JAK2 has a pivotal physiologic role in triggering several signaling pathways by binding to several cytokine receptors; therefore specific targeting of the mutant protein is absolutely required. Currently, no drug can discriminate between the mutant and the normal protein in myeloproliferative neoplasms. We have previously shown that an aromatic interaction between F617 and F594/F595 of pseudokinase domain helix C is required for constitutive signaling of JAK2 V617F, which constitutes the first molecular step in triggering hyperactivation in JAK2.
Aims
We aimed to find novel inhibitory strategies that directly destabilize the initial molecular step in V617F-mediated activation, namely the aromatic stacking between F617 and F594/F595 that only exist in the mutant conformation. We also aim to assess these approaches on the homologous JAK1 V658F, associated with ALL, and in the context of distinct cytokine receptors.
Methods
Via structure-guided mutagenesis, we assessed the role of key residues around the F617 aromatic interaction and used a combination of transcriptional activity luciferase-based assays, biochemical assays and cellular growth to measure the activity of JAKs in reconstituted JAK2 and JAK1 deficient cells.
Results
We created several aromatic mutations (S591F, A597F, A598F, S599F, V615F) strategically localizing around this aromatic interaction. We show that the specific mutation A598F preferentially inhibits JAK2 V617F over JAK2 WT, and without altering the normal function of the protein. Of importance, the inhibitory mutation A598F suppressed the hyperactivity of JAK2 V617F and other disease-associated JAK2 mutants such as JAK2 T875N and R683G, but not the JAK2 exon 12 mutant K539L.
We found that the mutation A598F and the homologous A639F can inhibit basal JAK2 V617F and JAK1 V658F, respectively, in the context of distinct cytokine receptors, such as IL2-R, IL9-R, IFNGR and type I IFN receptor (IFNAR).
Surprisingly, we found that the A598F mutation, as well as several other inhibitory mutations for JAK2 V617F, when in the background of the wild-type protein, drastically altered the cytokine responsiveness of the IFNGR, while it preserved normal cytokine response for EpoR, IL2-R, IL9R and IFNAR. This was even more compelling for JAK1 signaling with IFNGR; when these homologous inhibitory mutations were present in JAK1, they completely blocked the IFNγ signal transduction. Thus, it appears that the region of the JH2 helix C, which is required for JAK2 V617F and JAK1 V658F hyperactivation mechanism, is also crucial in specifically relaying the activation by cytokine in the heterodimeric IFNGR complex. Finally, we also show an unexpectedly low effect of JAK2 V617F on IFNGR complex compared to other cytokine receptors that physiologically utilize JAK2.
Conclusion
We identified a novel strategy to specifically inhibit JAK2 V617F and homologous JAK1 mutants, and we described an unexpected requirement for pseudokinase domain helix C specifically in the function of IFNGR. Inhibitory mutations localizing in the vicinity of JH2 helix aromatic interaction of JAK2 V617F restore normal function for most of JAK2-utilizing cytokine receptors except for the IFNGR.
Session topic: 15. Myeloproliferative neoplasms – Biology & Translational Research
Keyword(s): IFN-gamma, Janus Kinase inhibitor, Myeloproliferative disorder, Signal transduction