

Contributions
Abstract: S866
Type: Oral Presentation
Presentation during EHA23: On Saturday, June 16, 2018 from 17:00 - 17:15
Location: Room A4
Background
In acute lymphoblastic leukaemia (ALL) models, mesenchymal stromal cells (MSC) can form a ‘protective niche’ critical for treatment resistance (Ebinger, 2016; Duan, 2014).
Aims
We set out to determine the existence of such a niche in primary human bone marrow from patients with ALL and to understand the mechanism by which MSC may support ALL cell survival in this niche, following standard chemotherapy drugs.
Methods
We used both primary MSC and blasts from bone marrow of 152 patients enrolled on the UKALL14 trial as well as B-ALL cell lines and HS27a stromal cell line. We employed immunocytochemistry, gene expression profiling, ELISA, cytokine bead assay, MTS assays, apoptosis assays, quantification of reactive oxygen species (ROS), assays of mitochondrial number and various live cell imaging techniques.
Results
Immunostaining for f-actin, a gene expression profile panel and ELISAs showed that a significant proportion of primary patient bone marrow specimens contained MSC with an irreversible, activated phenotype, analagous to cancer associated fibroblasts (ALL-CAF). We demonstrated that both primary ALL cells, ALL cell lines and cytarabine (AraC) were able to generate ALL-CAF de novo from both healthy donor MSC and HS27a cells. ALL-CAF formation was closely associated with an increase in reactive oxygen species (ROS). It was functionally significant; pre-treatment of HS27a with AraC before co-culture with ALL cells led to increased cell proliferation and chemotherapy resistance. This phenomenon was not observed in untreated, or VCR or DEX pre-treated HS27a. Given AraC induces apoptosis by increasing ROS, we postulated MSC may protect ALL cells by ‘rescuing’ them from oxidative stress. We exposed SEM cells to AraC for 48 hours and observed a 1.4 fold increase in intracellular ROS (P=0.01), a 2-fold increase in apoptosis (p=0.0009) and cell death (p=0.01) at 48 hours compared to an untreated control. When SEM cells (a B-ALL cell line that didn't induce ALL-CAF) were co-cultured with MSC in the presence of AraC, ROS reduced 4-fold (p = 0.0002) compared to SEM in mono-culture which correlated with a 2.5-fold reduction in apoptosis (p=0.02) and a 3.4-fold reduction in cell death (p=0.0001). The observations couldn't be replicated in a transwell system, suggesting a contact-dependent mechanism. Next, using a mitotracker flow cytometry assay, we showed transfer of mitochondrial from HS27a to ALL cell lines, proportionate to the ROS level of the ALL cell line. Mitochondrial transfer to healthy donor B-cell controls was minimal. Passive transfer was excluded in a transwell system. Treatment of the MSC and ALL cell co-cultures with AraC increased mitochondrial transfer to ALL cells 3-fold at 72 hours compared to an untreated control (p <0.0001) and was AraC dose-dependent. Treatment with DEX and VCR did not impact mitochondrial transfer. We confirmed mitochondria were transferred along tunnelling nanotubes (TNT) using time-lapse confocal microscopy. As further evidence for transfer via TNT, the actin inhibitor latrunculin B and the microtubule damaging agent nocodazole both significantly blocked the phenomenon.
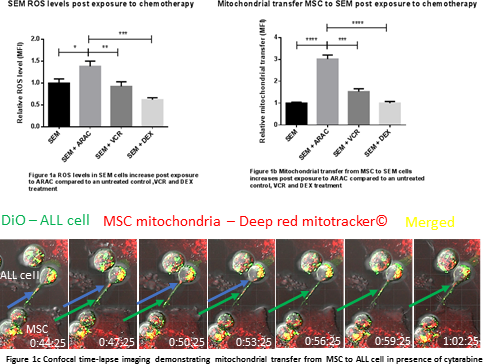
Conclusion
We have shown that CAF-like MSC can provide support to ALL cells under oxidative stress by mitochondrial transfer via TNT. Our data may explain the ineffectiveness of ROS-inducing chemotherapy at eradicating residual disease at this niche and provide an explanation of why low dose non-ROS-inducing, microtubule damaging agents such as VCR used in maintenance therapy are effective in ALL.
Session topic: 1. Acute lymphoblastic leukemia – Biology & Translational Research
Keyword(s): Acute lymphoblastic leukemia, Cytarabine, Microenvironment, Mitochondria
Abstract: S866
Type: Oral Presentation
Presentation during EHA23: On Saturday, June 16, 2018 from 17:00 - 17:15
Location: Room A4
Background
In acute lymphoblastic leukaemia (ALL) models, mesenchymal stromal cells (MSC) can form a ‘protective niche’ critical for treatment resistance (Ebinger, 2016; Duan, 2014).
Aims
We set out to determine the existence of such a niche in primary human bone marrow from patients with ALL and to understand the mechanism by which MSC may support ALL cell survival in this niche, following standard chemotherapy drugs.
Methods
We used both primary MSC and blasts from bone marrow of 152 patients enrolled on the UKALL14 trial as well as B-ALL cell lines and HS27a stromal cell line. We employed immunocytochemistry, gene expression profiling, ELISA, cytokine bead assay, MTS assays, apoptosis assays, quantification of reactive oxygen species (ROS), assays of mitochondrial number and various live cell imaging techniques.
Results
Immunostaining for f-actin, a gene expression profile panel and ELISAs showed that a significant proportion of primary patient bone marrow specimens contained MSC with an irreversible, activated phenotype, analagous to cancer associated fibroblasts (ALL-CAF). We demonstrated that both primary ALL cells, ALL cell lines and cytarabine (AraC) were able to generate ALL-CAF de novo from both healthy donor MSC and HS27a cells. ALL-CAF formation was closely associated with an increase in reactive oxygen species (ROS). It was functionally significant; pre-treatment of HS27a with AraC before co-culture with ALL cells led to increased cell proliferation and chemotherapy resistance. This phenomenon was not observed in untreated, or VCR or DEX pre-treated HS27a. Given AraC induces apoptosis by increasing ROS, we postulated MSC may protect ALL cells by ‘rescuing’ them from oxidative stress. We exposed SEM cells to AraC for 48 hours and observed a 1.4 fold increase in intracellular ROS (P=0.01), a 2-fold increase in apoptosis (p=0.0009) and cell death (p=0.01) at 48 hours compared to an untreated control. When SEM cells (a B-ALL cell line that didn't induce ALL-CAF) were co-cultured with MSC in the presence of AraC, ROS reduced 4-fold (p = 0.0002) compared to SEM in mono-culture which correlated with a 2.5-fold reduction in apoptosis (p=0.02) and a 3.4-fold reduction in cell death (p=0.0001). The observations couldn't be replicated in a transwell system, suggesting a contact-dependent mechanism. Next, using a mitotracker flow cytometry assay, we showed transfer of mitochondrial from HS27a to ALL cell lines, proportionate to the ROS level of the ALL cell line. Mitochondrial transfer to healthy donor B-cell controls was minimal. Passive transfer was excluded in a transwell system. Treatment of the MSC and ALL cell co-cultures with AraC increased mitochondrial transfer to ALL cells 3-fold at 72 hours compared to an untreated control (p <0.0001) and was AraC dose-dependent. Treatment with DEX and VCR did not impact mitochondrial transfer. We confirmed mitochondria were transferred along tunnelling nanotubes (TNT) using time-lapse confocal microscopy. As further evidence for transfer via TNT, the actin inhibitor latrunculin B and the microtubule damaging agent nocodazole both significantly blocked the phenomenon.
Conclusion
We have shown that CAF-like MSC can provide support to ALL cells under oxidative stress by mitochondrial transfer via TNT. Our data may explain the ineffectiveness of ROS-inducing chemotherapy at eradicating residual disease at this niche and provide an explanation of why low dose non-ROS-inducing, microtubule damaging agents such as VCR used in maintenance therapy are effective in ALL.
Session topic: 1. Acute lymphoblastic leukemia – Biology & Translational Research
Keyword(s): Acute lymphoblastic leukemia, Cytarabine, Microenvironment, Mitochondria