

Contributions
Abstract: S1570
Type: Oral Presentation
Presentation during EHA23: On Sunday, June 17, 2018 from 08:00 - 08:15
Location: Room A6
Background
Though numerous recurrent epigenetic mutations have been identified in chronic lymphocytic leukemia (CLL), their role in the molecular and clinical pathomechanism is only partly understood.
Aims
To provide the missing links between the cancer epigenotype and phenotype, we profiled the chromatin landscape and the transcriptome in primary peripheral blood cells of 19 CD19 sorted samples of untreated chronic lymphocytic leukemia (CLL) patients in comparison to 9 pools of non-malignant CD19-sorted B cells from 65 age-matched healthy donors.
Methods
We characterized DNA methylation by whole genome bisulfite sequencing, the major histone modification (H3K4me1, H3K4me3, H3K9me3, H3K9ac, H3K27me3, H3K27ac and H3K36me3) by ChIP-seq, nucleosome occupancy from high coverage H3 ChIP-seq and open chromatin sites were identified by the assay for transposase-accessible chromatin (ATAC-seq) in both bulk and single cells. In addition, RNA transcription was analyzed by strand-specific RNA-seq of both long and short RNAs.
Results
Remarkably, the CLL DNA methylome contains a large genome fraction of ~50% partially methylated domains in comparison to non-malignant B cells (<1 %). Auto- and cross-correlation of histone modifications showed changes for the active promoter mark H3K4me3 and for the repressive H3K27me3 modification, leading to terminal inactivation of poised promoters and activation of alternative transcriptional start sites in CLL. Most significant changes were observed at enhancers, and 256/279 (92%) of enhancers with changed H3K27ac signal that overlapped with partially methylated domains were activated. ATACseq uncovered 24400 differentially open loci at promoters, enhancers and repressed regions in CLL cells. Single cell ATACseq correlations of loci were used to infer spatial enhancer-promoter contacts and their local enrichment. In total 17,122 promoter-enhancer pairs were identified, with 11,585 promoters being assigned to different enhancers in CLL and non-malignant B cells. Of these re-wired promoters 921 and 438 were linked to genes down- and upregulated in CLL, respectively. We selected the most relevant transcription factors (TFs) that displayed differential binding in two or more of the chromatin readouts (Figure). The aberrant regulatory epigenetic signals were detected at 83% of the transcriptionally deregulated of genes in CLL, explaining the large majority of transcriptional dysregulation in CLL. The connections present in our B-cell gene regulatory network together with direct protein-protein interactions listed in the String database defined TF regulatory modules and provided links to chromatin modifiers. A gene set enrichment analysis of targets of these TFs retrieved NFkB and BCR signaling as the top hits. The network also includes the induction of the TCF4 in CLL via a large-scale enhancer activation and its linkage to expression of the anti-apototic BCL2 factor.
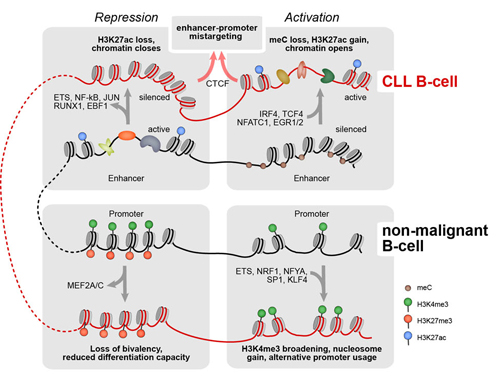
Conclusion
The deregulated TF network identified here rationalizes how the epigenetic pathophenotype of CLL is linked to changes in TF activity. Understanding the transcriptional dysregulation of the molecular targets of novel therapies will help to personalize treatment, reduce serious adverse events and thereby optimize outcome of CLL patients.
Session topic: 5. Chronic lymphocytic leukemia and related disorders – Biology & Translational Research
Keyword(s): BCL2, Chromatin structure, NF- B, NFAT
Abstract: S1570
Type: Oral Presentation
Presentation during EHA23: On Sunday, June 17, 2018 from 08:00 - 08:15
Location: Room A6
Background
Though numerous recurrent epigenetic mutations have been identified in chronic lymphocytic leukemia (CLL), their role in the molecular and clinical pathomechanism is only partly understood.
Aims
To provide the missing links between the cancer epigenotype and phenotype, we profiled the chromatin landscape and the transcriptome in primary peripheral blood cells of 19 CD19 sorted samples of untreated chronic lymphocytic leukemia (CLL) patients in comparison to 9 pools of non-malignant CD19-sorted B cells from 65 age-matched healthy donors.
Methods
We characterized DNA methylation by whole genome bisulfite sequencing, the major histone modification (H3K4me1, H3K4me3, H3K9me3, H3K9ac, H3K27me3, H3K27ac and H3K36me3) by ChIP-seq, nucleosome occupancy from high coverage H3 ChIP-seq and open chromatin sites were identified by the assay for transposase-accessible chromatin (ATAC-seq) in both bulk and single cells. In addition, RNA transcription was analyzed by strand-specific RNA-seq of both long and short RNAs.
Results
Remarkably, the CLL DNA methylome contains a large genome fraction of ~50% partially methylated domains in comparison to non-malignant B cells (<1 %). Auto- and cross-correlation of histone modifications showed changes for the active promoter mark H3K4me3 and for the repressive H3K27me3 modification, leading to terminal inactivation of poised promoters and activation of alternative transcriptional start sites in CLL. Most significant changes were observed at enhancers, and 256/279 (92%) of enhancers with changed H3K27ac signal that overlapped with partially methylated domains were activated. ATACseq uncovered 24400 differentially open loci at promoters, enhancers and repressed regions in CLL cells. Single cell ATACseq correlations of loci were used to infer spatial enhancer-promoter contacts and their local enrichment. In total 17,122 promoter-enhancer pairs were identified, with 11,585 promoters being assigned to different enhancers in CLL and non-malignant B cells. Of these re-wired promoters 921 and 438 were linked to genes down- and upregulated in CLL, respectively. We selected the most relevant transcription factors (TFs) that displayed differential binding in two or more of the chromatin readouts (Figure). The aberrant regulatory epigenetic signals were detected at 83% of the transcriptionally deregulated of genes in CLL, explaining the large majority of transcriptional dysregulation in CLL. The connections present in our B-cell gene regulatory network together with direct protein-protein interactions listed in the String database defined TF regulatory modules and provided links to chromatin modifiers. A gene set enrichment analysis of targets of these TFs retrieved NFkB and BCR signaling as the top hits. The network also includes the induction of the TCF4 in CLL via a large-scale enhancer activation and its linkage to expression of the anti-apototic BCL2 factor.
Conclusion
The deregulated TF network identified here rationalizes how the epigenetic pathophenotype of CLL is linked to changes in TF activity. Understanding the transcriptional dysregulation of the molecular targets of novel therapies will help to personalize treatment, reduce serious adverse events and thereby optimize outcome of CLL patients.
Session topic: 5. Chronic lymphocytic leukemia and related disorders – Biology & Translational Research
Keyword(s): BCL2, Chromatin structure, NF- B, NFAT