

Contributions
Abstract: S831
Type: Oral Presentation
Presentation during EHA23: On Saturday, June 16, 2018 from 12:30 - 12:45
Location: Room A7
Background
Acquired ibrutinib resistance due to BTKCys481 mutations occurs in B-cell malignancies, including those with MYD88 mutations. BTKCys481 mutations are usually sub-clonal, and their relevance to clinical progression remains unclear.
Aims
We sought to define the signaling pathways that promote ibrutinib resistance in MYD88 mutated WM and ABC DLBCL cells. Furthermore, we sought to delineate mechanisms responsible for promoting ibrutinib resistance in neighboring BTK wild-type tumor cells.
Methods
We engineered BTKCys481Ser and BTKWT expressing MYD88 mutated WM and ABC DLBCL cells, and performed signaling studies that included pathways responsible for aberrant MYD88 signaling, and downstream BTK signaling. Furthermore, cytokine release was evaluated by multiplex assay and correlated to findings in patients who progressed on ibrutinib after acquisition of BTK Cys 481 mutations. Lastly, co-culture experiments with BTK Cys 481 mutated and unmutated cells were performed and viability assessed.
Results
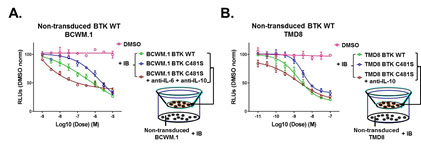
Comparing to BTKWT, BTKCys481Ser expressing cells re-activates BTK–PLCγ2–ERK1/2 signaling in the presence of ibrutinib. Use of ERK1/2 inhibitors triggered apoptosis in BTKCys481Ser expressing cells, and showed synergistic cytotoxicity with ibrutinib. ERK1/2 re-activation in ibrutinib treated BTKCys481Ser cells was accompanied by release of many pro-survival and inflammatory cytokines, including IL-6 and IL-10 that were also blocked by ERK1/2 inhibition. To clarify if cytokine release by ibrutinib treated BTKCys481Ser cells could protect non-transduced BTKWT MYD88 mutated malignant cells, we used a TranswellTM co-culture system, and showed that non-transduced BTKWT MYD88 mutated WM or ABC DLBCL cells were rescued from ibrutinib induced cell death when co-cultured with BTKCys481Ser but not their BTKWT expressing counterparts (Figure 1). Use of IL-6 and/or IL-10 blocking antibodies abolished the protective effect conferred on non-transduced BTKWT by co-culture with BTKCys481Ser expressing WM or ABC DLBCL cell counterparts. Rebound of IL-6 and IL-10 serum levels also accompanied disease progression in WM patients with acquired BTKCys481 mutations.
Conclusion
Our findings show that the BTKCys481Ser mutation drives ibrutinib resistance in MYD88 mutated WM and ABC DLBCL cells through re-activation of ERK1/2 activation, and can confer a protective effect on BTKWT cells through a paracrine mechanism. The findings provide evidence for interclonal tumor cooperativity in mediating ibrutinib resistance.
Session topic: 19. Non-Hodgkin lymphoma Biology & Translational Research
Keyword(s): B cell lymphoma, ibrutinib, Resistance
Abstract: S831
Type: Oral Presentation
Presentation during EHA23: On Saturday, June 16, 2018 from 12:30 - 12:45
Location: Room A7
Background
Acquired ibrutinib resistance due to BTKCys481 mutations occurs in B-cell malignancies, including those with MYD88 mutations. BTKCys481 mutations are usually sub-clonal, and their relevance to clinical progression remains unclear.
Aims
We sought to define the signaling pathways that promote ibrutinib resistance in MYD88 mutated WM and ABC DLBCL cells. Furthermore, we sought to delineate mechanisms responsible for promoting ibrutinib resistance in neighboring BTK wild-type tumor cells.
Methods
We engineered BTKCys481Ser and BTKWT expressing MYD88 mutated WM and ABC DLBCL cells, and performed signaling studies that included pathways responsible for aberrant MYD88 signaling, and downstream BTK signaling. Furthermore, cytokine release was evaluated by multiplex assay and correlated to findings in patients who progressed on ibrutinib after acquisition of BTK Cys 481 mutations. Lastly, co-culture experiments with BTK Cys 481 mutated and unmutated cells were performed and viability assessed.
Results
Comparing to BTKWT, BTKCys481Ser expressing cells re-activates BTK–PLCγ2–ERK1/2 signaling in the presence of ibrutinib. Use of ERK1/2 inhibitors triggered apoptosis in BTKCys481Ser expressing cells, and showed synergistic cytotoxicity with ibrutinib. ERK1/2 re-activation in ibrutinib treated BTKCys481Ser cells was accompanied by release of many pro-survival and inflammatory cytokines, including IL-6 and IL-10 that were also blocked by ERK1/2 inhibition. To clarify if cytokine release by ibrutinib treated BTKCys481Ser cells could protect non-transduced BTKWT MYD88 mutated malignant cells, we used a TranswellTM co-culture system, and showed that non-transduced BTKWT MYD88 mutated WM or ABC DLBCL cells were rescued from ibrutinib induced cell death when co-cultured with BTKCys481Ser but not their BTKWT expressing counterparts (Figure 1). Use of IL-6 and/or IL-10 blocking antibodies abolished the protective effect conferred on non-transduced BTKWT by co-culture with BTKCys481Ser expressing WM or ABC DLBCL cell counterparts. Rebound of IL-6 and IL-10 serum levels also accompanied disease progression in WM patients with acquired BTKCys481 mutations.
Conclusion
Our findings show that the BTKCys481Ser mutation drives ibrutinib resistance in MYD88 mutated WM and ABC DLBCL cells through re-activation of ERK1/2 activation, and can confer a protective effect on BTKWT cells through a paracrine mechanism. The findings provide evidence for interclonal tumor cooperativity in mediating ibrutinib resistance.
Session topic: 19. Non-Hodgkin lymphoma Biology & Translational Research
Keyword(s): B cell lymphoma, ibrutinib, Resistance