

Contributions
Abstract: S437
Type: Oral Presentation
Presentation during EHA22: On Saturday, June 24, 2017 from 11:45 - 12:00
Location: Room N105
Background
Persistence of residual leukemia cells, due to deficiencies in apoptotic programs, is a major driver of relapse. Activation of alternative non-apoptotic cell death pathways such as necroptosis represents an attractive strategy to eliminate residual leukemia cells and prevent relapse. We have previously shown that SMAC-mimetics (SM) potently induce cell death by simultaneous RIP1-dependent apoptosis and necroptosis in a subset of refractory acute lymphoblastic leukemia (B-ALL) patient-derived samples. The molecular signals that drive sensitivity to RIP1-dependent cell death remained elusive so far.
Aims
The aim of this project was to understand the mechanisms that determine the specific vulnerability to necroptosis in ALL.
Methods
To identify molecular determinants of sensitivity to SM, we correlated the gene expression profiles of 17 primary samples with high and low sensitivity to SM with the IC50 in response to two SM compounds, birinapant and LCL161. We confirmed the top scoring genes including TNF receptor 1 (TNFR1) and TNFR2 by quantitative RT-PCR in patient-derived xenografts. We further validated our results by quantifying the expression of the candidate genes in an independent cohort of relapsed primary B-ALL and by screening samples with different expression levels of TNFR1 and 2 for their response to SM in vitro.
Results
Comparative gene expression profiling indicated a correlation of the expression of TNFR2 with sensitivity to SM in primary ALL. Using an independent cohort of relapsed ALL samples, we found that high TNFR2 expression predicted sensitivity to SM in an ex vivo model of the bone marrow. Deletion of either TNFR1 or TNFR2 using CRISPR/Cas9 in patient-derived ALL conferred resistance to treatment with SM in vivo in the xenograft model, indicating that TNFR1 and 2 are both functionally required for cell death. In agreement with an important role for TNFR2 in the response to SM, the overexpression of TNFR2 leads to increased sensitivity to the TNFR1/RIP1 death axis. On the mechanistic level, recruitment of RIP1 to TNFR1 is a key event in the activation of cell death, which is abolished in TNFR2-deficient leukemia and does not occur in SM resistant cases.
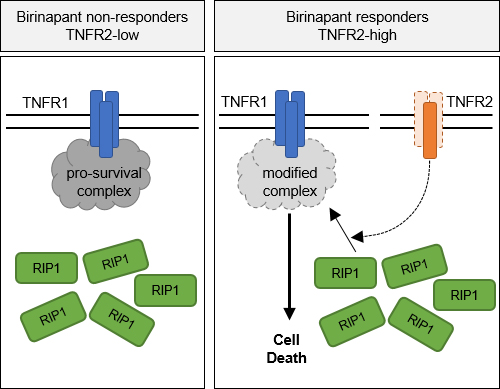
Conclusion
Taken together, our data reveal a novel function of TNFR2 in cell death signaling, as TNFR2 predicts sensitivity to SMAC mimetics and plays a key role in activating the TNFR1/RIP1 cell death pathway, which underlies the switch from RIP1-controlled cell survival to cell death and characterizes a distinct vulnerability in ALL.
Session topic: 1. Acute lymphoblastic leukemia - Biology
Keyword(s): Tumor necrosis factor (TNF), Acute lymphoblastic leukemia
Abstract: S437
Type: Oral Presentation
Presentation during EHA22: On Saturday, June 24, 2017 from 11:45 - 12:00
Location: Room N105
Background
Persistence of residual leukemia cells, due to deficiencies in apoptotic programs, is a major driver of relapse. Activation of alternative non-apoptotic cell death pathways such as necroptosis represents an attractive strategy to eliminate residual leukemia cells and prevent relapse. We have previously shown that SMAC-mimetics (SM) potently induce cell death by simultaneous RIP1-dependent apoptosis and necroptosis in a subset of refractory acute lymphoblastic leukemia (B-ALL) patient-derived samples. The molecular signals that drive sensitivity to RIP1-dependent cell death remained elusive so far.
Aims
The aim of this project was to understand the mechanisms that determine the specific vulnerability to necroptosis in ALL.
Methods
To identify molecular determinants of sensitivity to SM, we correlated the gene expression profiles of 17 primary samples with high and low sensitivity to SM with the IC50 in response to two SM compounds, birinapant and LCL161. We confirmed the top scoring genes including TNF receptor 1 (TNFR1) and TNFR2 by quantitative RT-PCR in patient-derived xenografts. We further validated our results by quantifying the expression of the candidate genes in an independent cohort of relapsed primary B-ALL and by screening samples with different expression levels of TNFR1 and 2 for their response to SM in vitro.
Results
Comparative gene expression profiling indicated a correlation of the expression of TNFR2 with sensitivity to SM in primary ALL. Using an independent cohort of relapsed ALL samples, we found that high TNFR2 expression predicted sensitivity to SM in an ex vivo model of the bone marrow. Deletion of either TNFR1 or TNFR2 using CRISPR/Cas9 in patient-derived ALL conferred resistance to treatment with SM in vivo in the xenograft model, indicating that TNFR1 and 2 are both functionally required for cell death. In agreement with an important role for TNFR2 in the response to SM, the overexpression of TNFR2 leads to increased sensitivity to the TNFR1/RIP1 death axis. On the mechanistic level, recruitment of RIP1 to TNFR1 is a key event in the activation of cell death, which is abolished in TNFR2-deficient leukemia and does not occur in SM resistant cases.
Conclusion
Taken together, our data reveal a novel function of TNFR2 in cell death signaling, as TNFR2 predicts sensitivity to SMAC mimetics and plays a key role in activating the TNFR1/RIP1 cell death pathway, which underlies the switch from RIP1-controlled cell survival to cell death and characterizes a distinct vulnerability in ALL.
Session topic: 1. Acute lymphoblastic leukemia - Biology
Keyword(s): Tumor necrosis factor (TNF), Acute lymphoblastic leukemia