

Contributions
Abstract: S120
Type: Oral Presentation
Presentation during EHA22: On Friday, June 23, 2017 from 11:45 - 12:00
Location: Hall E
Background
Clinical heterogeneity of myelodysplastic syndromes (MDS) and related myeloid neoplasms reflects molecular diversity. Most common genetic associations with distinct clinical or pathomorphologic phenotypes have been already reported, but many other common somatic lesions exist and their clinical context still remains elusive. AT rich interactive domain 2 (ARID2), which is located on chromosome 12q, encodes a component of the SWI/SNF complex that is involved in chromatin remodeling. In recent years multiple groups detected ARID2 mutations in a variety of solid tumors.
Aims
Here, we present whole exome sequencing-guided identification of novel ARID2 mutations in myeloid neoplasms. Specifically, in addition to copy number analysis and deep targeted and exome sequencing, here we include RNA sequencing and splicing analyses of the roles of spliceosomal mutations in ARID2 missplicing and gene expression.
Methods
Bone marrow aspirates or blood samples were collected from 1,473 patients with MDS (n=455), myelodysplastic/myeloproliferative neoplasms (MDS/MPN) (n=201), myeloproliferative neoplasms (MPN) (n=56), sAML (n=221), and primary acute myeloid leukemia (pAML) (n=540) at the Cleveland Clinic and The University of Tokyo; the registered data at The Cancer Genome Atlas were also included. Diagnoses were classified using World Health Organization criteria. Informed consent for sample collection was obtained according to a protocol approved by each Institutional Review Board in accordance with the Declaration of Helsinki.
Results
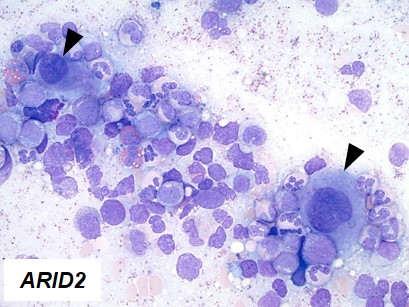
By comprehensive genetic investigation of these cases, we characterized here cases (10%) in which decreased expression of ARID2 mediated their clinical effects in MDS and other myeloid neoplasms via multiple kinds of genetic lesions. We showed that insufficient ARID2 expression mainly in MDS arose from ARID2 mutations, deletions, and missplicing due to U2AF1 mutations that yielded defective ARID2 transcripts. Clonal architecture analyses showed that ARID2 mutations and deletions occurred as initial events of MDS or myelodysplastic/myeloproliferative neoplasms, and not during progression to acute myeloid leukemia. Morphologically, progressive maturation in myeloid and erythroid lineages and hypolobated megakaryocytes (indicated by arrow heads in Figure 1) were common in cases with ARID2 mutations and deletions, and were also found in cases with U2AF1 mutations. Functionally, we utilized in vitro knockdown models of ARID2 expression in hematopoietic cell lines and bone marrow mononuclear cells. Since no homozygous deletion or mutation of ARID2 was identified, we transduced shRNA in neoplastic and healthy hematopoietic cells to obtain disease models with partial reduction of ARID2 expression. Two myeloid cell lines (HL60 and K562) in which ARID2 expression was knocked down showed significantly lower cell counts compared to those with normal ARID2 expression, compatible with more apoptotic cells in knockdown experiments. Flow cytometric analysis of the cell lines with reduced ARID2 expression revealed increased cell-surface maturation markers, CD11b and glycophorin A (GPA), suggesting that reduced expression of ARID2 resulted in more differentiation in myeloid and erythroid lineages. Knockdown of ARID2 failed to reduce colony formation in bone marrow mononuclear cells. These results indicate that reduced ARID2 expression might induce more differentiation in myeloid/erythroid lineages and more apoptosis to reduce cell populations without reduction of proliferation capacity in hematopoietic progenitor cells. Finally, we examined morphological findings associated with knockdown ARID2 expression. Compared to control cells, K562 cells with reduced ARID2 expression formed more hypolobated megakaryocytes, which confirmed morphological findings seen in ARID2 and U2AF1 defects.
Conclusion
ARID2 is a MDS-suppressor gene whose expression is attenuated by multiple mechanisms as it shapes the distinct morphological phenotype of a subset of myelodysplasia.
Session topic: 9. Myelodysplastic syndromes - Biology
Keyword(s): Myelodysplasia, Mutation status, Megakaryocyte, MDS
Abstract: S120
Type: Oral Presentation
Presentation during EHA22: On Friday, June 23, 2017 from 11:45 - 12:00
Location: Hall E
Background
Clinical heterogeneity of myelodysplastic syndromes (MDS) and related myeloid neoplasms reflects molecular diversity. Most common genetic associations with distinct clinical or pathomorphologic phenotypes have been already reported, but many other common somatic lesions exist and their clinical context still remains elusive. AT rich interactive domain 2 (ARID2), which is located on chromosome 12q, encodes a component of the SWI/SNF complex that is involved in chromatin remodeling. In recent years multiple groups detected ARID2 mutations in a variety of solid tumors.
Aims
Here, we present whole exome sequencing-guided identification of novel ARID2 mutations in myeloid neoplasms. Specifically, in addition to copy number analysis and deep targeted and exome sequencing, here we include RNA sequencing and splicing analyses of the roles of spliceosomal mutations in ARID2 missplicing and gene expression.
Methods
Bone marrow aspirates or blood samples were collected from 1,473 patients with MDS (n=455), myelodysplastic/myeloproliferative neoplasms (MDS/MPN) (n=201), myeloproliferative neoplasms (MPN) (n=56), sAML (n=221), and primary acute myeloid leukemia (pAML) (n=540) at the Cleveland Clinic and The University of Tokyo; the registered data at The Cancer Genome Atlas were also included. Diagnoses were classified using World Health Organization criteria. Informed consent for sample collection was obtained according to a protocol approved by each Institutional Review Board in accordance with the Declaration of Helsinki.
Results
By comprehensive genetic investigation of these cases, we characterized here cases (10%) in which decreased expression of ARID2 mediated their clinical effects in MDS and other myeloid neoplasms via multiple kinds of genetic lesions. We showed that insufficient ARID2 expression mainly in MDS arose from ARID2 mutations, deletions, and missplicing due to U2AF1 mutations that yielded defective ARID2 transcripts. Clonal architecture analyses showed that ARID2 mutations and deletions occurred as initial events of MDS or myelodysplastic/myeloproliferative neoplasms, and not during progression to acute myeloid leukemia. Morphologically, progressive maturation in myeloid and erythroid lineages and hypolobated megakaryocytes (indicated by arrow heads in Figure 1) were common in cases with ARID2 mutations and deletions, and were also found in cases with U2AF1 mutations. Functionally, we utilized in vitro knockdown models of ARID2 expression in hematopoietic cell lines and bone marrow mononuclear cells. Since no homozygous deletion or mutation of ARID2 was identified, we transduced shRNA in neoplastic and healthy hematopoietic cells to obtain disease models with partial reduction of ARID2 expression. Two myeloid cell lines (HL60 and K562) in which ARID2 expression was knocked down showed significantly lower cell counts compared to those with normal ARID2 expression, compatible with more apoptotic cells in knockdown experiments. Flow cytometric analysis of the cell lines with reduced ARID2 expression revealed increased cell-surface maturation markers, CD11b and glycophorin A (GPA), suggesting that reduced expression of ARID2 resulted in more differentiation in myeloid and erythroid lineages. Knockdown of ARID2 failed to reduce colony formation in bone marrow mononuclear cells. These results indicate that reduced ARID2 expression might induce more differentiation in myeloid/erythroid lineages and more apoptosis to reduce cell populations without reduction of proliferation capacity in hematopoietic progenitor cells. Finally, we examined morphological findings associated with knockdown ARID2 expression. Compared to control cells, K562 cells with reduced ARID2 expression formed more hypolobated megakaryocytes, which confirmed morphological findings seen in ARID2 and U2AF1 defects.
Conclusion
ARID2 is a MDS-suppressor gene whose expression is attenuated by multiple mechanisms as it shapes the distinct morphological phenotype of a subset of myelodysplasia.
Session topic: 9. Myelodysplastic syndromes - Biology
Keyword(s): Myelodysplasia, Mutation status, Megakaryocyte, MDS