
IDENTIFICATION OF CARRIERS OF GENETIC ALTERATIONS IN THE HBB GLOBIN CLUSTER: HEMATIMETRIC AND BIOCHEMICAL DATA IN 462 HETEROZYGOTE INDIVIDUALS
(Abstract release date: 05/19/16)
EHA Library. Nieto J. 06/09/16; 135020; PB2120

Dr. Jorge M. Nieto
Contributions
Contributions
Abstract
Abstract: PB2120
Type: Publication Only
Background
The β globin cluster (HBB cluster) contains several genes differentially expressed over development. Most common alterations in this cluster are point mutations in the β globin gene (βº, β+ or β++ mutations depending on the severity) that produce β thalassemia. Additionally, large deletions of one or more genes can occur in the HBB cluster, leading to β like thalassemias (εγδβ, δβ or β thalassemia) or Hereditary Persistence of Fetal Hemoglobin (HPFH). Homozygotes or compound heterozygotes for HBB cluster alterations present variable transfusional requirements, depending on the combination of inherited alterations. In many centers, identification of carriers of these alterations is commonly done at hematimetric and biochemical level. Thus, effective identification of carriers and discrimination between different types of HBB alterations at this level is necessary for an appropriate genetic counseling.
Aims
To describe the hematimetric and biochemical profile of individuals bearing heterozygous alterations in the HBB cluster in order to facilitate their identification.
Methods
Large deletions and β gene point mutations were identified by Multiplex Ligation dependent Probe Amplification (MLPA) and sequencing, respectively. Most common alterations in the HBA globin cluster were assessed with the Alpha-Globin StripAssay. Hematimetric data were obtained with a Coulter LH 750 analyzer. HbF and HbA2 levels were obtained by ion exchange HPLC. In line with literature, individuals were categorized according to the type of deletion (εγδβº, δβº, HPFH or βº) or point mutation (βº, β+ or β++) in the HBB cluster. Coinheritance of alterations in the HBA globin cluster was a reason for exclusion. 462 carriers of alterations in the HBB cluster were finally included in the study.
Results
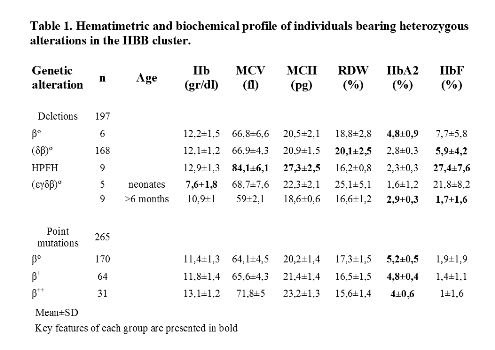
Means and standard deviations for hematimetric data variables, HbF and HbA2 levels for different HBB alterations are summarized in Table 1.
Conclusion
Within deletions, only βº deletions have elevated HbA2 levels (≥3,5%). The main differences between δβ thalassemia and HPFH were HbF levels, MCV and RDW. εγδβ thalassemia cases had variable features in relation to age; severe microcytic anemia was observed in neonates (4 out of 5 required transfusions) and marked microcytosis with mild anemia in adults. Many εγδβ thalassemias are caused by de novo deletions, so this condition should be suspected when severe microcytic anemia occurs in neonates, even if there is no history of familiar microcytosis.Roughly, 10% of δβ thalassemias have normal HbF (≤2%) and mimic an α thalassemia phenotype (microcytic and hypocromic red cells, normal HbA2 and HbF levels). When an alpha thalassemia trait is present, elevated RDW (≥16) makes us to suspect that the patient is actually a carrier of a δβº deletion.When a point mutation is present, phenotype aggravates accordingly to β globin production impairment. Generally, these individuals can be easily identified because of elevated HBA2 levels.In conclusion, phenotypes associated with alterations in the HBB cluster are frequently distinctive, although phenotypic overlap can be seen in a subset of cases and molecular analysis may be required.
Session topic: E-poster
Keyword(s): Genetic, Globin locus, Phenotype, Thalassemia
Type: Publication Only
Background
The β globin cluster (HBB cluster) contains several genes differentially expressed over development. Most common alterations in this cluster are point mutations in the β globin gene (βº, β+ or β++ mutations depending on the severity) that produce β thalassemia. Additionally, large deletions of one or more genes can occur in the HBB cluster, leading to β like thalassemias (εγδβ, δβ or β thalassemia) or Hereditary Persistence of Fetal Hemoglobin (HPFH). Homozygotes or compound heterozygotes for HBB cluster alterations present variable transfusional requirements, depending on the combination of inherited alterations. In many centers, identification of carriers of these alterations is commonly done at hematimetric and biochemical level. Thus, effective identification of carriers and discrimination between different types of HBB alterations at this level is necessary for an appropriate genetic counseling.
Aims
To describe the hematimetric and biochemical profile of individuals bearing heterozygous alterations in the HBB cluster in order to facilitate their identification.
Methods
Large deletions and β gene point mutations were identified by Multiplex Ligation dependent Probe Amplification (MLPA) and sequencing, respectively. Most common alterations in the HBA globin cluster were assessed with the Alpha-Globin StripAssay. Hematimetric data were obtained with a Coulter LH 750 analyzer. HbF and HbA2 levels were obtained by ion exchange HPLC. In line with literature, individuals were categorized according to the type of deletion (εγδβº, δβº, HPFH or βº) or point mutation (βº, β+ or β++) in the HBB cluster. Coinheritance of alterations in the HBA globin cluster was a reason for exclusion. 462 carriers of alterations in the HBB cluster were finally included in the study.
Results
Means and standard deviations for hematimetric data variables, HbF and HbA2 levels for different HBB alterations are summarized in Table 1.
Conclusion
Within deletions, only βº deletions have elevated HbA2 levels (≥3,5%). The main differences between δβ thalassemia and HPFH were HbF levels, MCV and RDW. εγδβ thalassemia cases had variable features in relation to age; severe microcytic anemia was observed in neonates (4 out of 5 required transfusions) and marked microcytosis with mild anemia in adults. Many εγδβ thalassemias are caused by de novo deletions, so this condition should be suspected when severe microcytic anemia occurs in neonates, even if there is no history of familiar microcytosis.Roughly, 10% of δβ thalassemias have normal HbF (≤2%) and mimic an α thalassemia phenotype (microcytic and hypocromic red cells, normal HbA2 and HbF levels). When an alpha thalassemia trait is present, elevated RDW (≥16) makes us to suspect that the patient is actually a carrier of a δβº deletion.When a point mutation is present, phenotype aggravates accordingly to β globin production impairment. Generally, these individuals can be easily identified because of elevated HBA2 levels.In conclusion, phenotypes associated with alterations in the HBB cluster are frequently distinctive, although phenotypic overlap can be seen in a subset of cases and molecular analysis may be required.
Session topic: E-poster
Keyword(s): Genetic, Globin locus, Phenotype, Thalassemia
Abstract: PB2120
Type: Publication Only
Background
The β globin cluster (HBB cluster) contains several genes differentially expressed over development. Most common alterations in this cluster are point mutations in the β globin gene (βº, β+ or β++ mutations depending on the severity) that produce β thalassemia. Additionally, large deletions of one or more genes can occur in the HBB cluster, leading to β like thalassemias (εγδβ, δβ or β thalassemia) or Hereditary Persistence of Fetal Hemoglobin (HPFH). Homozygotes or compound heterozygotes for HBB cluster alterations present variable transfusional requirements, depending on the combination of inherited alterations. In many centers, identification of carriers of these alterations is commonly done at hematimetric and biochemical level. Thus, effective identification of carriers and discrimination between different types of HBB alterations at this level is necessary for an appropriate genetic counseling.
Aims
To describe the hematimetric and biochemical profile of individuals bearing heterozygous alterations in the HBB cluster in order to facilitate their identification.
Methods
Large deletions and β gene point mutations were identified by Multiplex Ligation dependent Probe Amplification (MLPA) and sequencing, respectively. Most common alterations in the HBA globin cluster were assessed with the Alpha-Globin StripAssay. Hematimetric data were obtained with a Coulter LH 750 analyzer. HbF and HbA2 levels were obtained by ion exchange HPLC. In line with literature, individuals were categorized according to the type of deletion (εγδβº, δβº, HPFH or βº) or point mutation (βº, β+ or β++) in the HBB cluster. Coinheritance of alterations in the HBA globin cluster was a reason for exclusion. 462 carriers of alterations in the HBB cluster were finally included in the study.
Results
Means and standard deviations for hematimetric data variables, HbF and HbA2 levels for different HBB alterations are summarized in Table 1.
Conclusion
Within deletions, only βº deletions have elevated HbA2 levels (≥3,5%). The main differences between δβ thalassemia and HPFH were HbF levels, MCV and RDW. εγδβ thalassemia cases had variable features in relation to age; severe microcytic anemia was observed in neonates (4 out of 5 required transfusions) and marked microcytosis with mild anemia in adults. Many εγδβ thalassemias are caused by de novo deletions, so this condition should be suspected when severe microcytic anemia occurs in neonates, even if there is no history of familiar microcytosis.Roughly, 10% of δβ thalassemias have normal HbF (≤2%) and mimic an α thalassemia phenotype (microcytic and hypocromic red cells, normal HbA2 and HbF levels). When an alpha thalassemia trait is present, elevated RDW (≥16) makes us to suspect that the patient is actually a carrier of a δβº deletion.When a point mutation is present, phenotype aggravates accordingly to β globin production impairment. Generally, these individuals can be easily identified because of elevated HBA2 levels.In conclusion, phenotypes associated with alterations in the HBB cluster are frequently distinctive, although phenotypic overlap can be seen in a subset of cases and molecular analysis may be required.
Session topic: E-poster
Keyword(s): Genetic, Globin locus, Phenotype, Thalassemia
Type: Publication Only
Background
The β globin cluster (HBB cluster) contains several genes differentially expressed over development. Most common alterations in this cluster are point mutations in the β globin gene (βº, β+ or β++ mutations depending on the severity) that produce β thalassemia. Additionally, large deletions of one or more genes can occur in the HBB cluster, leading to β like thalassemias (εγδβ, δβ or β thalassemia) or Hereditary Persistence of Fetal Hemoglobin (HPFH). Homozygotes or compound heterozygotes for HBB cluster alterations present variable transfusional requirements, depending on the combination of inherited alterations. In many centers, identification of carriers of these alterations is commonly done at hematimetric and biochemical level. Thus, effective identification of carriers and discrimination between different types of HBB alterations at this level is necessary for an appropriate genetic counseling.
Aims
To describe the hematimetric and biochemical profile of individuals bearing heterozygous alterations in the HBB cluster in order to facilitate their identification.
Methods
Large deletions and β gene point mutations were identified by Multiplex Ligation dependent Probe Amplification (MLPA) and sequencing, respectively. Most common alterations in the HBA globin cluster were assessed with the Alpha-Globin StripAssay. Hematimetric data were obtained with a Coulter LH 750 analyzer. HbF and HbA2 levels were obtained by ion exchange HPLC. In line with literature, individuals were categorized according to the type of deletion (εγδβº, δβº, HPFH or βº) or point mutation (βº, β+ or β++) in the HBB cluster. Coinheritance of alterations in the HBA globin cluster was a reason for exclusion. 462 carriers of alterations in the HBB cluster were finally included in the study.
Results
Means and standard deviations for hematimetric data variables, HbF and HbA2 levels for different HBB alterations are summarized in Table 1.
Conclusion
Within deletions, only βº deletions have elevated HbA2 levels (≥3,5%). The main differences between δβ thalassemia and HPFH were HbF levels, MCV and RDW. εγδβ thalassemia cases had variable features in relation to age; severe microcytic anemia was observed in neonates (4 out of 5 required transfusions) and marked microcytosis with mild anemia in adults. Many εγδβ thalassemias are caused by de novo deletions, so this condition should be suspected when severe microcytic anemia occurs in neonates, even if there is no history of familiar microcytosis.Roughly, 10% of δβ thalassemias have normal HbF (≤2%) and mimic an α thalassemia phenotype (microcytic and hypocromic red cells, normal HbA2 and HbF levels). When an alpha thalassemia trait is present, elevated RDW (≥16) makes us to suspect that the patient is actually a carrier of a δβº deletion.When a point mutation is present, phenotype aggravates accordingly to β globin production impairment. Generally, these individuals can be easily identified because of elevated HBA2 levels.In conclusion, phenotypes associated with alterations in the HBB cluster are frequently distinctive, although phenotypic overlap can be seen in a subset of cases and molecular analysis may be required.
Session topic: E-poster
Keyword(s): Genetic, Globin locus, Phenotype, Thalassemia
{{ help_message }}
{{filter}}