
HEMOPHAGOCYTIC LYMPHOHISTIOCYTOSIS: EXPERIENCE OF AN HOSPITAL IN NORTHEN SPAIN IN THE LAST 22 YEARS
(Abstract release date: 05/19/16)
EHA Library. Martinez-Robles V. 06/09/16; 134955; PB2055

Dr. Violeta Martinez-Robles
Contributions
Contributions
Abstract
Abstract: PB2055
Type: Publication Only
Background
Hemophagocytic lymphohistiocytosis (HLH) is a very unusual and disastrous dysfunction of the immune system leading to an uncontrolled cytokine storm that has become more recognized over the past decade. HLH was first described in 1939 by Scott and Robb-Smith.This disease is characterized by lengthy and overwhelming activation of antigen-presenting cells (macrophages, histiocytes) and CD8+ T and NK cells. All this scenario produces an important hyperinflammatory condition and organ damage including fever, splenomegaly, cytopenias, coagulophaty and/or hypertrigliceridemia. Histiocyte Society (HS) criteria have been extensively applied for diagnosing HLH, however not all of them are usually showed at the initial presentation. HLH could be displayed in two different contexts: primary (genetic, usually in children, known as familial form) and secondary (acquired), caused by malignancy, infections, metabolic conditions or autoimmune disorders. This disorder is a life-threatening disease which should be suspected and treated as soon as possible.
Aims
Analyzing the casuistry of hemophagocytic syndromes, diagnostic criteria, treatment applied and evolution in our hospital and making a comparison with the current literature.
Methods
A retrospective analysis was carried out through the medical records of all patients with suspected diagnosis of HLH between 1994 and 2016 in one hospital. Age, clinical features, diagnostic criteria proposed by the HS, etiology, treatment and evolution were analyzed. In our study only 17 out of 49 patients met the requested criteria.
Results
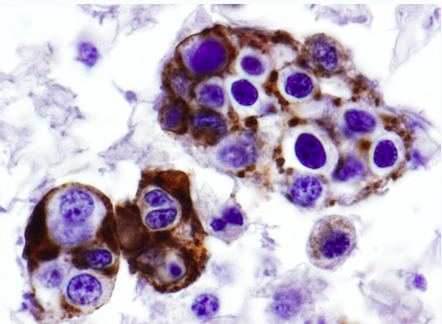
The number of cases was 17 (M7/F10). The median age at diagnosis was 67 years, ranged between 4 and 84 years. Clinical development was fast in almost all patients, except in two of them with long-standing pancytopenia. The most frequent causes of consultation were fever and general syndrome. All of them met 5 or more of the criteria necessary for the diagnosis. sIL-2R level and NK activity were underestimated because they were not tested in more than half of the cases. All of them were secondary (11 malignancies, 4 infections, 1 autoimmune disease), unable to clarify the cause in 1 of them. Only 8 patients survived HLH (47%), although 6 died for other complications. 94/04 HLH treatment according to protocol was established in 5 of them. In the rest of them the triggering cause was treated. The most frequent causes of death were liver failure, infectious complications and bleeding.Test results were: hyperferritinemia (100%), hypertriglyceridemia (87.5%), liver enzyme alteration (52.9%), hypofibrinogenemia (33.3%), esplenomegaly (70.6%), hemophagocytosis in bone marrow (94.1%), 100% of tested patients presented high sIL-2R level.
Conclusion
Despite being a serious disease and with rapidly torpid course it remains underdiagnosed, obtaining the diagnosis, in most of the cases after hemophagocytosis phenomena is visualized in bone marrow. According to the literature, the main reasons for consultation and hospitalization are similar to those presented in our hospital. The response to treatment is difficult to compare because most of patients has been treated with different therapeutic regimens. In conclusion, it is always needed considering the possibility of a HLH diagnosis when we have a patient with fever and pancytopenia not clearly explained, and the importance of requesting simple test with high profitability such as ferritin and triglycerides in all suggestive picture.
Session topic: E-poster
Keyword(s): Fever, Immune response, NK cell, Pancytopenia
Type: Publication Only
Background
Hemophagocytic lymphohistiocytosis (HLH) is a very unusual and disastrous dysfunction of the immune system leading to an uncontrolled cytokine storm that has become more recognized over the past decade. HLH was first described in 1939 by Scott and Robb-Smith.This disease is characterized by lengthy and overwhelming activation of antigen-presenting cells (macrophages, histiocytes) and CD8+ T and NK cells. All this scenario produces an important hyperinflammatory condition and organ damage including fever, splenomegaly, cytopenias, coagulophaty and/or hypertrigliceridemia. Histiocyte Society (HS) criteria have been extensively applied for diagnosing HLH, however not all of them are usually showed at the initial presentation. HLH could be displayed in two different contexts: primary (genetic, usually in children, known as familial form) and secondary (acquired), caused by malignancy, infections, metabolic conditions or autoimmune disorders. This disorder is a life-threatening disease which should be suspected and treated as soon as possible.
Aims
Analyzing the casuistry of hemophagocytic syndromes, diagnostic criteria, treatment applied and evolution in our hospital and making a comparison with the current literature.
Methods
A retrospective analysis was carried out through the medical records of all patients with suspected diagnosis of HLH between 1994 and 2016 in one hospital. Age, clinical features, diagnostic criteria proposed by the HS, etiology, treatment and evolution were analyzed. In our study only 17 out of 49 patients met the requested criteria.
Results
The number of cases was 17 (M7/F10). The median age at diagnosis was 67 years, ranged between 4 and 84 years. Clinical development was fast in almost all patients, except in two of them with long-standing pancytopenia. The most frequent causes of consultation were fever and general syndrome. All of them met 5 or more of the criteria necessary for the diagnosis. sIL-2R level and NK activity were underestimated because they were not tested in more than half of the cases. All of them were secondary (11 malignancies, 4 infections, 1 autoimmune disease), unable to clarify the cause in 1 of them. Only 8 patients survived HLH (47%), although 6 died for other complications. 94/04 HLH treatment according to protocol was established in 5 of them. In the rest of them the triggering cause was treated. The most frequent causes of death were liver failure, infectious complications and bleeding.Test results were: hyperferritinemia (100%), hypertriglyceridemia (87.5%), liver enzyme alteration (52.9%), hypofibrinogenemia (33.3%), esplenomegaly (70.6%), hemophagocytosis in bone marrow (94.1%), 100% of tested patients presented high sIL-2R level.
Conclusion
Despite being a serious disease and with rapidly torpid course it remains underdiagnosed, obtaining the diagnosis, in most of the cases after hemophagocytosis phenomena is visualized in bone marrow. According to the literature, the main reasons for consultation and hospitalization are similar to those presented in our hospital. The response to treatment is difficult to compare because most of patients has been treated with different therapeutic regimens. In conclusion, it is always needed considering the possibility of a HLH diagnosis when we have a patient with fever and pancytopenia not clearly explained, and the importance of requesting simple test with high profitability such as ferritin and triglycerides in all suggestive picture.
Session topic: E-poster
Keyword(s): Fever, Immune response, NK cell, Pancytopenia
Abstract: PB2055
Type: Publication Only
Background
Hemophagocytic lymphohistiocytosis (HLH) is a very unusual and disastrous dysfunction of the immune system leading to an uncontrolled cytokine storm that has become more recognized over the past decade. HLH was first described in 1939 by Scott and Robb-Smith.This disease is characterized by lengthy and overwhelming activation of antigen-presenting cells (macrophages, histiocytes) and CD8+ T and NK cells. All this scenario produces an important hyperinflammatory condition and organ damage including fever, splenomegaly, cytopenias, coagulophaty and/or hypertrigliceridemia. Histiocyte Society (HS) criteria have been extensively applied for diagnosing HLH, however not all of them are usually showed at the initial presentation. HLH could be displayed in two different contexts: primary (genetic, usually in children, known as familial form) and secondary (acquired), caused by malignancy, infections, metabolic conditions or autoimmune disorders. This disorder is a life-threatening disease which should be suspected and treated as soon as possible.
Aims
Analyzing the casuistry of hemophagocytic syndromes, diagnostic criteria, treatment applied and evolution in our hospital and making a comparison with the current literature.
Methods
A retrospective analysis was carried out through the medical records of all patients with suspected diagnosis of HLH between 1994 and 2016 in one hospital. Age, clinical features, diagnostic criteria proposed by the HS, etiology, treatment and evolution were analyzed. In our study only 17 out of 49 patients met the requested criteria.
Results
The number of cases was 17 (M7/F10). The median age at diagnosis was 67 years, ranged between 4 and 84 years. Clinical development was fast in almost all patients, except in two of them with long-standing pancytopenia. The most frequent causes of consultation were fever and general syndrome. All of them met 5 or more of the criteria necessary for the diagnosis. sIL-2R level and NK activity were underestimated because they were not tested in more than half of the cases. All of them were secondary (11 malignancies, 4 infections, 1 autoimmune disease), unable to clarify the cause in 1 of them. Only 8 patients survived HLH (47%), although 6 died for other complications. 94/04 HLH treatment according to protocol was established in 5 of them. In the rest of them the triggering cause was treated. The most frequent causes of death were liver failure, infectious complications and bleeding.Test results were: hyperferritinemia (100%), hypertriglyceridemia (87.5%), liver enzyme alteration (52.9%), hypofibrinogenemia (33.3%), esplenomegaly (70.6%), hemophagocytosis in bone marrow (94.1%), 100% of tested patients presented high sIL-2R level.
Conclusion
Despite being a serious disease and with rapidly torpid course it remains underdiagnosed, obtaining the diagnosis, in most of the cases after hemophagocytosis phenomena is visualized in bone marrow. According to the literature, the main reasons for consultation and hospitalization are similar to those presented in our hospital. The response to treatment is difficult to compare because most of patients has been treated with different therapeutic regimens. In conclusion, it is always needed considering the possibility of a HLH diagnosis when we have a patient with fever and pancytopenia not clearly explained, and the importance of requesting simple test with high profitability such as ferritin and triglycerides in all suggestive picture.
Session topic: E-poster
Keyword(s): Fever, Immune response, NK cell, Pancytopenia
Type: Publication Only
Background
Hemophagocytic lymphohistiocytosis (HLH) is a very unusual and disastrous dysfunction of the immune system leading to an uncontrolled cytokine storm that has become more recognized over the past decade. HLH was first described in 1939 by Scott and Robb-Smith.This disease is characterized by lengthy and overwhelming activation of antigen-presenting cells (macrophages, histiocytes) and CD8+ T and NK cells. All this scenario produces an important hyperinflammatory condition and organ damage including fever, splenomegaly, cytopenias, coagulophaty and/or hypertrigliceridemia. Histiocyte Society (HS) criteria have been extensively applied for diagnosing HLH, however not all of them are usually showed at the initial presentation. HLH could be displayed in two different contexts: primary (genetic, usually in children, known as familial form) and secondary (acquired), caused by malignancy, infections, metabolic conditions or autoimmune disorders. This disorder is a life-threatening disease which should be suspected and treated as soon as possible.
Aims
Analyzing the casuistry of hemophagocytic syndromes, diagnostic criteria, treatment applied and evolution in our hospital and making a comparison with the current literature.
Methods
A retrospective analysis was carried out through the medical records of all patients with suspected diagnosis of HLH between 1994 and 2016 in one hospital. Age, clinical features, diagnostic criteria proposed by the HS, etiology, treatment and evolution were analyzed. In our study only 17 out of 49 patients met the requested criteria.
Results
The number of cases was 17 (M7/F10). The median age at diagnosis was 67 years, ranged between 4 and 84 years. Clinical development was fast in almost all patients, except in two of them with long-standing pancytopenia. The most frequent causes of consultation were fever and general syndrome. All of them met 5 or more of the criteria necessary for the diagnosis. sIL-2R level and NK activity were underestimated because they were not tested in more than half of the cases. All of them were secondary (11 malignancies, 4 infections, 1 autoimmune disease), unable to clarify the cause in 1 of them. Only 8 patients survived HLH (47%), although 6 died for other complications. 94/04 HLH treatment according to protocol was established in 5 of them. In the rest of them the triggering cause was treated. The most frequent causes of death were liver failure, infectious complications and bleeding.Test results were: hyperferritinemia (100%), hypertriglyceridemia (87.5%), liver enzyme alteration (52.9%), hypofibrinogenemia (33.3%), esplenomegaly (70.6%), hemophagocytosis in bone marrow (94.1%), 100% of tested patients presented high sIL-2R level.
Conclusion
Despite being a serious disease and with rapidly torpid course it remains underdiagnosed, obtaining the diagnosis, in most of the cases after hemophagocytosis phenomena is visualized in bone marrow. According to the literature, the main reasons for consultation and hospitalization are similar to those presented in our hospital. The response to treatment is difficult to compare because most of patients has been treated with different therapeutic regimens. In conclusion, it is always needed considering the possibility of a HLH diagnosis when we have a patient with fever and pancytopenia not clearly explained, and the importance of requesting simple test with high profitability such as ferritin and triglycerides in all suggestive picture.
Session topic: E-poster
Keyword(s): Fever, Immune response, NK cell, Pancytopenia
{{ help_message }}
{{filter}}