
TECHNICAL VALIDATION OF A NEXT-GENERATION SEQUENCING PANEL FOR ACUTE MYELOID LEUKEMIA DIAGNOSIS
(Abstract release date: 05/19/16)
EHA Library. Llop M. 06/09/16; 134524; PB1624

Dr. Marta Llop
Contributions
Contributions
Abstract
Abstract: PB1624
Type: Publication Only
Background
Recurrently mutated genes have been described for acute myeloid leukemia (AML). However, only a few (NPM1, CEBPA, FLT3) have prognostic and clinical implications and are systematically analysed, usually by individual assays. The development of high throughput techniques as next-generation sequencing (NGS) allows a parallel detection of these markers and enables the inclusion of novel molecular targets.
Aims
Our aim is to validate the clinical applicability to routine laboratories of a hotspot NGS panel that includes recurrently validated mutated genes and other potentially actionable targets.
Methods
We included 130 de novo AML samples previously characterized for FLT3-D835, NPM1-T288, DNMT3A-R882, and CEBPA mutations by conventional molecular biology techniques (CMBT). We tested the Ion Ampliseq AML community panel (Life Technologies) (IAACP) which requires 40 ng of DNA per sample and includes hotspots of ASXL1, BRAF, CBL, FLT3, IDH1, IDH2, JAK 2, KIT, KRAS, NRAS, PTPN11, RUNX1 and WT1; and the entire coding sequence of CEBPA, DNMT3A, GATA2, TET2 and TP53. The design does not include the FLT3-ITD region. Libraries were sequenced in the Ion PGM (n=12) and the Ion Proton platforms (n=118). For comparison, 3 patients were sequenced in both platforms. To determine if the hotspot approach is adequate, a subset of 25 patients was also analyzed with a custom panel (Sure Design Tool (Agilent)) that included the complete coding sequence of the genes included in the IAACP except from CBL and GATA2. Libraries were prepared with 250 ng of DNA per sample and sequenced in an Illumina platform.
Results
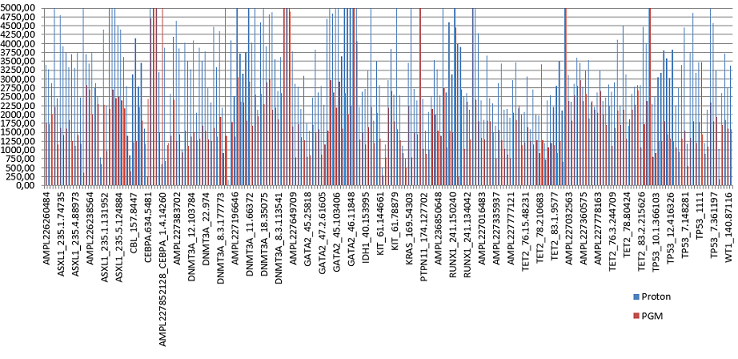
The IAACP sequenced in the Proton platform yielded a mean depth of 5916 reads, mean uniformity was 92.19% and mean on-target reads 89.05%. Two amplicons (RUNX1_239.66909 and ASXL1_235.1.76662) showed a strong strand bias (x5) towards the forward strand systematically. The PGM platform yielded a mean depth of 2500 reads, mean uniformity of 91.86% and mean on-target reads of 93.39%. GATA2_47.1.16110 and DNMT3A_23.66955 amplicons showed a strong bias towards the forward strand (100X) and the reverse strand (42X), respectively. For both platforms we observed intra-run differences regarding read depth and strand bias, however, none of the 239 amplicons showed a mean depth lower that 250X (Figure 1). Patients sequenced in both platforms yielded the same mutations.IAACP found 100% of the previously known mutations by CMBT plus 11 extra mutations that were negative by CMBT. These mutations were reconfirmed by Sanger sequencing.Among the 25 patients analysed with the custom panel, 53 variants were found. Fifty (95%) were also detected with the IAACP. The remaining 3 variants (5%) were located outside the hotspot regions in genes with poor clinical significance. Additionally, the IAACP detected 13 variants in the overlapping regions not found by the custom panel.
Conclusion
In conclusion, the IAACP is reliable and reproducible for routine diagnosis. It allows detection of known important mutations with increased sensibility compared with CMBT, and also detection of complementary mutations with potential clinical implications. The main limitation of its design is the absence of amplicons covering FLT3-ITD region. The hotspot approach yields a higher mean read depth than the whole coding sequence analysis without significant losses and it requires less input DNA. Moreover, analysis of strand bias and low coverage regions is mandatory to assess sequencing quality and to perform extra assays if necessary. Funding PI13/01640 and PIE13/00046.
Session topic: E-poster
Keyword(s): Acute myeloid leukemia, Diagnosis, Molecular markers, Mutation analysis
Type: Publication Only
Background
Recurrently mutated genes have been described for acute myeloid leukemia (AML). However, only a few (NPM1, CEBPA, FLT3) have prognostic and clinical implications and are systematically analysed, usually by individual assays. The development of high throughput techniques as next-generation sequencing (NGS) allows a parallel detection of these markers and enables the inclusion of novel molecular targets.
Aims
Our aim is to validate the clinical applicability to routine laboratories of a hotspot NGS panel that includes recurrently validated mutated genes and other potentially actionable targets.
Methods
We included 130 de novo AML samples previously characterized for FLT3-D835, NPM1-T288, DNMT3A-R882, and CEBPA mutations by conventional molecular biology techniques (CMBT). We tested the Ion Ampliseq AML community panel (Life Technologies) (IAACP) which requires 40 ng of DNA per sample and includes hotspots of ASXL1, BRAF, CBL, FLT3, IDH1, IDH2, JAK 2, KIT, KRAS, NRAS, PTPN11, RUNX1 and WT1; and the entire coding sequence of CEBPA, DNMT3A, GATA2, TET2 and TP53. The design does not include the FLT3-ITD region. Libraries were sequenced in the Ion PGM (n=12) and the Ion Proton platforms (n=118). For comparison, 3 patients were sequenced in both platforms. To determine if the hotspot approach is adequate, a subset of 25 patients was also analyzed with a custom panel (Sure Design Tool (Agilent)) that included the complete coding sequence of the genes included in the IAACP except from CBL and GATA2. Libraries were prepared with 250 ng of DNA per sample and sequenced in an Illumina platform.
Results
The IAACP sequenced in the Proton platform yielded a mean depth of 5916 reads, mean uniformity was 92.19% and mean on-target reads 89.05%. Two amplicons (RUNX1_239.66909 and ASXL1_235.1.76662) showed a strong strand bias (x5) towards the forward strand systematically. The PGM platform yielded a mean depth of 2500 reads, mean uniformity of 91.86% and mean on-target reads of 93.39%. GATA2_47.1.16110 and DNMT3A_23.66955 amplicons showed a strong bias towards the forward strand (100X) and the reverse strand (42X), respectively. For both platforms we observed intra-run differences regarding read depth and strand bias, however, none of the 239 amplicons showed a mean depth lower that 250X (Figure 1). Patients sequenced in both platforms yielded the same mutations.IAACP found 100% of the previously known mutations by CMBT plus 11 extra mutations that were negative by CMBT. These mutations were reconfirmed by Sanger sequencing.Among the 25 patients analysed with the custom panel, 53 variants were found. Fifty (95%) were also detected with the IAACP. The remaining 3 variants (5%) were located outside the hotspot regions in genes with poor clinical significance. Additionally, the IAACP detected 13 variants in the overlapping regions not found by the custom panel.
Conclusion
In conclusion, the IAACP is reliable and reproducible for routine diagnosis. It allows detection of known important mutations with increased sensibility compared with CMBT, and also detection of complementary mutations with potential clinical implications. The main limitation of its design is the absence of amplicons covering FLT3-ITD region. The hotspot approach yields a higher mean read depth than the whole coding sequence analysis without significant losses and it requires less input DNA. Moreover, analysis of strand bias and low coverage regions is mandatory to assess sequencing quality and to perform extra assays if necessary. Funding PI13/01640 and PIE13/00046.
Session topic: E-poster
Keyword(s): Acute myeloid leukemia, Diagnosis, Molecular markers, Mutation analysis
Abstract: PB1624
Type: Publication Only
Background
Recurrently mutated genes have been described for acute myeloid leukemia (AML). However, only a few (NPM1, CEBPA, FLT3) have prognostic and clinical implications and are systematically analysed, usually by individual assays. The development of high throughput techniques as next-generation sequencing (NGS) allows a parallel detection of these markers and enables the inclusion of novel molecular targets.
Aims
Our aim is to validate the clinical applicability to routine laboratories of a hotspot NGS panel that includes recurrently validated mutated genes and other potentially actionable targets.
Methods
We included 130 de novo AML samples previously characterized for FLT3-D835, NPM1-T288, DNMT3A-R882, and CEBPA mutations by conventional molecular biology techniques (CMBT). We tested the Ion Ampliseq AML community panel (Life Technologies) (IAACP) which requires 40 ng of DNA per sample and includes hotspots of ASXL1, BRAF, CBL, FLT3, IDH1, IDH2, JAK 2, KIT, KRAS, NRAS, PTPN11, RUNX1 and WT1; and the entire coding sequence of CEBPA, DNMT3A, GATA2, TET2 and TP53. The design does not include the FLT3-ITD region. Libraries were sequenced in the Ion PGM (n=12) and the Ion Proton platforms (n=118). For comparison, 3 patients were sequenced in both platforms. To determine if the hotspot approach is adequate, a subset of 25 patients was also analyzed with a custom panel (Sure Design Tool (Agilent)) that included the complete coding sequence of the genes included in the IAACP except from CBL and GATA2. Libraries were prepared with 250 ng of DNA per sample and sequenced in an Illumina platform.
Results
The IAACP sequenced in the Proton platform yielded a mean depth of 5916 reads, mean uniformity was 92.19% and mean on-target reads 89.05%. Two amplicons (RUNX1_239.66909 and ASXL1_235.1.76662) showed a strong strand bias (x5) towards the forward strand systematically. The PGM platform yielded a mean depth of 2500 reads, mean uniformity of 91.86% and mean on-target reads of 93.39%. GATA2_47.1.16110 and DNMT3A_23.66955 amplicons showed a strong bias towards the forward strand (100X) and the reverse strand (42X), respectively. For both platforms we observed intra-run differences regarding read depth and strand bias, however, none of the 239 amplicons showed a mean depth lower that 250X (Figure 1). Patients sequenced in both platforms yielded the same mutations.IAACP found 100% of the previously known mutations by CMBT plus 11 extra mutations that were negative by CMBT. These mutations were reconfirmed by Sanger sequencing.Among the 25 patients analysed with the custom panel, 53 variants were found. Fifty (95%) were also detected with the IAACP. The remaining 3 variants (5%) were located outside the hotspot regions in genes with poor clinical significance. Additionally, the IAACP detected 13 variants in the overlapping regions not found by the custom panel.
Conclusion
In conclusion, the IAACP is reliable and reproducible for routine diagnosis. It allows detection of known important mutations with increased sensibility compared with CMBT, and also detection of complementary mutations with potential clinical implications. The main limitation of its design is the absence of amplicons covering FLT3-ITD region. The hotspot approach yields a higher mean read depth than the whole coding sequence analysis without significant losses and it requires less input DNA. Moreover, analysis of strand bias and low coverage regions is mandatory to assess sequencing quality and to perform extra assays if necessary. Funding PI13/01640 and PIE13/00046.
Session topic: E-poster
Keyword(s): Acute myeloid leukemia, Diagnosis, Molecular markers, Mutation analysis
Type: Publication Only
Background
Recurrently mutated genes have been described for acute myeloid leukemia (AML). However, only a few (NPM1, CEBPA, FLT3) have prognostic and clinical implications and are systematically analysed, usually by individual assays. The development of high throughput techniques as next-generation sequencing (NGS) allows a parallel detection of these markers and enables the inclusion of novel molecular targets.
Aims
Our aim is to validate the clinical applicability to routine laboratories of a hotspot NGS panel that includes recurrently validated mutated genes and other potentially actionable targets.
Methods
We included 130 de novo AML samples previously characterized for FLT3-D835, NPM1-T288, DNMT3A-R882, and CEBPA mutations by conventional molecular biology techniques (CMBT). We tested the Ion Ampliseq AML community panel (Life Technologies) (IAACP) which requires 40 ng of DNA per sample and includes hotspots of ASXL1, BRAF, CBL, FLT3, IDH1, IDH2, JAK 2, KIT, KRAS, NRAS, PTPN11, RUNX1 and WT1; and the entire coding sequence of CEBPA, DNMT3A, GATA2, TET2 and TP53. The design does not include the FLT3-ITD region. Libraries were sequenced in the Ion PGM (n=12) and the Ion Proton platforms (n=118). For comparison, 3 patients were sequenced in both platforms. To determine if the hotspot approach is adequate, a subset of 25 patients was also analyzed with a custom panel (Sure Design Tool (Agilent)) that included the complete coding sequence of the genes included in the IAACP except from CBL and GATA2. Libraries were prepared with 250 ng of DNA per sample and sequenced in an Illumina platform.
Results
The IAACP sequenced in the Proton platform yielded a mean depth of 5916 reads, mean uniformity was 92.19% and mean on-target reads 89.05%. Two amplicons (RUNX1_239.66909 and ASXL1_235.1.76662) showed a strong strand bias (x5) towards the forward strand systematically. The PGM platform yielded a mean depth of 2500 reads, mean uniformity of 91.86% and mean on-target reads of 93.39%. GATA2_47.1.16110 and DNMT3A_23.66955 amplicons showed a strong bias towards the forward strand (100X) and the reverse strand (42X), respectively. For both platforms we observed intra-run differences regarding read depth and strand bias, however, none of the 239 amplicons showed a mean depth lower that 250X (Figure 1). Patients sequenced in both platforms yielded the same mutations.IAACP found 100% of the previously known mutations by CMBT plus 11 extra mutations that were negative by CMBT. These mutations were reconfirmed by Sanger sequencing.Among the 25 patients analysed with the custom panel, 53 variants were found. Fifty (95%) were also detected with the IAACP. The remaining 3 variants (5%) were located outside the hotspot regions in genes with poor clinical significance. Additionally, the IAACP detected 13 variants in the overlapping regions not found by the custom panel.
Conclusion
In conclusion, the IAACP is reliable and reproducible for routine diagnosis. It allows detection of known important mutations with increased sensibility compared with CMBT, and also detection of complementary mutations with potential clinical implications. The main limitation of its design is the absence of amplicons covering FLT3-ITD region. The hotspot approach yields a higher mean read depth than the whole coding sequence analysis without significant losses and it requires less input DNA. Moreover, analysis of strand bias and low coverage regions is mandatory to assess sequencing quality and to perform extra assays if necessary. Funding PI13/01640 and PIE13/00046.
Session topic: E-poster
Keyword(s): Acute myeloid leukemia, Diagnosis, Molecular markers, Mutation analysis
{{ help_message }}
{{filter}}