
“SEPARATING THE WHEAT FROM THE CHAFF” – CONGENITAL HEMOLYTIC ANEMIA STUDY WITH A TARGETED NEXT GENERATION SEQUENCING PANEL
(Abstract release date: 05/19/16)
EHA Library. Magalhães Maia T. 06/09/16; 133037; E1488

Tabita Magalhães Maia
Contributions
Contributions
Abstract
Abstract: E1488
Type: Eposter Presentation
Background
Diagnosing a Coombs negative hemolytic anemia (HA) can be a true challenge, specially in the absence of family history or when the peripheral blood smear is not very informative. A few years ago, we had to sequence gene by gene by Sanger methodology, which was time costly. Nowadays, with Next Generation Sequencing (NGS) panels it is much faster, nevertheless we may be drowned in a large number of putative pathological mutations. The concept of one mutation (or two for the autossomal recessive disorders) one disease, have changed and now we have to deal with pathological variations or not, or modulating factors. As a reference laboratory dedicated to the study of benign red blood cell disorders (RBC) we have many of HAs with an identified molecular lesion, but also several cases, that even though thoroughly screened for hemoglobinopathies, enzymes and membrane defects remained without identification of the pathological mutations. This has implications at prognostic and therapeutic level, and makes the correct genetic counseling impossible.
Aims
Identify the molecular causes in a group of 14 patients with congenital HA, after screening for the most common hemoglobinopathies, enzymopathies and membranopathies.
Methods
Informed consent was signed for all the individuals in the study. We have used a targeted NGS panel encompassing the exonic regions of 24 genes (del Orbe et al, 2015) and sequenced 14 samples from patients with a congenital HA. In all the samples the common screening tests for RBC disorders were preformed, like high performance liquid chromatography (HPLC) for Hb variants, enzymatic quantification and membrane disorders screening tests as cryohemolysis test and eosin-5'-maleimide . The variants found were classified using in sillico analysis and the presence of pathological and likely pathogenic variants were confirmed by Sanger sequencing and, when available, familial studies were performed.
Results
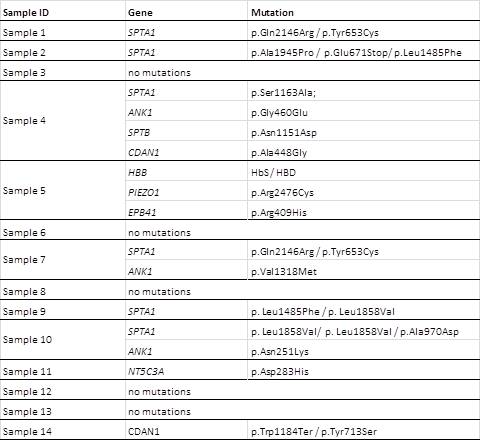
All the samples showed a great number of variants, the majority were benign or likely benign (a media of 320/sample) or of unknown significance or likely pathogenic (media of 25/sample). Only 8 samples showed pathologic variants that can justify the clinical presentation. Most of them are associated with membrane disorders (Table). In 5 of the samples no pathological mutations were found, in one sample (sample 11) we found a NT5C3A missense mutation but in the heterozygous state (Sanger sequencing of all gene also didn’t reveal any other mutation). Three samples are compound heterozygous for mutations in the SPTA1 gene, three have a combination of mutations in different genes affecting membrane structure, one is a compound heterozygous for two missense CDAN1 mutations not described before. Sample 5 is form a sickle cell disease patient with an uncommon presentation in the newborn period, a PIEZO1 mutation inherited from the mother and a EPB41 mutation inherited from the father are likely to be the causes of the aggravated phenotype.Table – Results of the NGS sequencing – pathological variants
Conclusion
After in sillico analysis of the variants found by NGS (almost 350 per sample) and confirmation by Sanger sequencing of the pathological variants, we have identified a molecular cause for the HA in 8 of the 14 samples studied.This study confirms that target NGS is the most complete and cheaper method for sequencing the genes associated with AH but, for the cases that remain without mutations, whole exome sequencing or whole genome sequencing need to be done, in order to identify new genes that can later be added to the AH panel.
Session topic: E-poster
Keyword(s): Genotype, Hemolytic anemia
Type: Eposter Presentation
Background
Diagnosing a Coombs negative hemolytic anemia (HA) can be a true challenge, specially in the absence of family history or when the peripheral blood smear is not very informative. A few years ago, we had to sequence gene by gene by Sanger methodology, which was time costly. Nowadays, with Next Generation Sequencing (NGS) panels it is much faster, nevertheless we may be drowned in a large number of putative pathological mutations. The concept of one mutation (or two for the autossomal recessive disorders) one disease, have changed and now we have to deal with pathological variations or not, or modulating factors. As a reference laboratory dedicated to the study of benign red blood cell disorders (RBC) we have many of HAs with an identified molecular lesion, but also several cases, that even though thoroughly screened for hemoglobinopathies, enzymes and membrane defects remained without identification of the pathological mutations. This has implications at prognostic and therapeutic level, and makes the correct genetic counseling impossible.
Aims
Identify the molecular causes in a group of 14 patients with congenital HA, after screening for the most common hemoglobinopathies, enzymopathies and membranopathies.
Methods
Informed consent was signed for all the individuals in the study. We have used a targeted NGS panel encompassing the exonic regions of 24 genes (del Orbe et al, 2015) and sequenced 14 samples from patients with a congenital HA. In all the samples the common screening tests for RBC disorders were preformed, like high performance liquid chromatography (HPLC) for Hb variants, enzymatic quantification and membrane disorders screening tests as cryohemolysis test and eosin-5'-maleimide . The variants found were classified using in sillico analysis and the presence of pathological and likely pathogenic variants were confirmed by Sanger sequencing and, when available, familial studies were performed.
Results
All the samples showed a great number of variants, the majority were benign or likely benign (a media of 320/sample) or of unknown significance or likely pathogenic (media of 25/sample). Only 8 samples showed pathologic variants that can justify the clinical presentation. Most of them are associated with membrane disorders (Table). In 5 of the samples no pathological mutations were found, in one sample (sample 11) we found a NT5C3A missense mutation but in the heterozygous state (Sanger sequencing of all gene also didn’t reveal any other mutation). Three samples are compound heterozygous for mutations in the SPTA1 gene, three have a combination of mutations in different genes affecting membrane structure, one is a compound heterozygous for two missense CDAN1 mutations not described before. Sample 5 is form a sickle cell disease patient with an uncommon presentation in the newborn period, a PIEZO1 mutation inherited from the mother and a EPB41 mutation inherited from the father are likely to be the causes of the aggravated phenotype.Table – Results of the NGS sequencing – pathological variants
Conclusion
After in sillico analysis of the variants found by NGS (almost 350 per sample) and confirmation by Sanger sequencing of the pathological variants, we have identified a molecular cause for the HA in 8 of the 14 samples studied.This study confirms that target NGS is the most complete and cheaper method for sequencing the genes associated with AH but, for the cases that remain without mutations, whole exome sequencing or whole genome sequencing need to be done, in order to identify new genes that can later be added to the AH panel.
Session topic: E-poster
Keyword(s): Genotype, Hemolytic anemia
Abstract: E1488
Type: Eposter Presentation
Background
Diagnosing a Coombs negative hemolytic anemia (HA) can be a true challenge, specially in the absence of family history or when the peripheral blood smear is not very informative. A few years ago, we had to sequence gene by gene by Sanger methodology, which was time costly. Nowadays, with Next Generation Sequencing (NGS) panels it is much faster, nevertheless we may be drowned in a large number of putative pathological mutations. The concept of one mutation (or two for the autossomal recessive disorders) one disease, have changed and now we have to deal with pathological variations or not, or modulating factors. As a reference laboratory dedicated to the study of benign red blood cell disorders (RBC) we have many of HAs with an identified molecular lesion, but also several cases, that even though thoroughly screened for hemoglobinopathies, enzymes and membrane defects remained without identification of the pathological mutations. This has implications at prognostic and therapeutic level, and makes the correct genetic counseling impossible.
Aims
Identify the molecular causes in a group of 14 patients with congenital HA, after screening for the most common hemoglobinopathies, enzymopathies and membranopathies.
Methods
Informed consent was signed for all the individuals in the study. We have used a targeted NGS panel encompassing the exonic regions of 24 genes (del Orbe et al, 2015) and sequenced 14 samples from patients with a congenital HA. In all the samples the common screening tests for RBC disorders were preformed, like high performance liquid chromatography (HPLC) for Hb variants, enzymatic quantification and membrane disorders screening tests as cryohemolysis test and eosin-5'-maleimide . The variants found were classified using in sillico analysis and the presence of pathological and likely pathogenic variants were confirmed by Sanger sequencing and, when available, familial studies were performed.
Results
All the samples showed a great number of variants, the majority were benign or likely benign (a media of 320/sample) or of unknown significance or likely pathogenic (media of 25/sample). Only 8 samples showed pathologic variants that can justify the clinical presentation. Most of them are associated with membrane disorders (Table). In 5 of the samples no pathological mutations were found, in one sample (sample 11) we found a NT5C3A missense mutation but in the heterozygous state (Sanger sequencing of all gene also didn’t reveal any other mutation). Three samples are compound heterozygous for mutations in the SPTA1 gene, three have a combination of mutations in different genes affecting membrane structure, one is a compound heterozygous for two missense CDAN1 mutations not described before. Sample 5 is form a sickle cell disease patient with an uncommon presentation in the newborn period, a PIEZO1 mutation inherited from the mother and a EPB41 mutation inherited from the father are likely to be the causes of the aggravated phenotype.Table – Results of the NGS sequencing – pathological variants
Conclusion
After in sillico analysis of the variants found by NGS (almost 350 per sample) and confirmation by Sanger sequencing of the pathological variants, we have identified a molecular cause for the HA in 8 of the 14 samples studied.This study confirms that target NGS is the most complete and cheaper method for sequencing the genes associated with AH but, for the cases that remain without mutations, whole exome sequencing or whole genome sequencing need to be done, in order to identify new genes that can later be added to the AH panel.
Session topic: E-poster
Keyword(s): Genotype, Hemolytic anemia
Type: Eposter Presentation
Background
Diagnosing a Coombs negative hemolytic anemia (HA) can be a true challenge, specially in the absence of family history or when the peripheral blood smear is not very informative. A few years ago, we had to sequence gene by gene by Sanger methodology, which was time costly. Nowadays, with Next Generation Sequencing (NGS) panels it is much faster, nevertheless we may be drowned in a large number of putative pathological mutations. The concept of one mutation (or two for the autossomal recessive disorders) one disease, have changed and now we have to deal with pathological variations or not, or modulating factors. As a reference laboratory dedicated to the study of benign red blood cell disorders (RBC) we have many of HAs with an identified molecular lesion, but also several cases, that even though thoroughly screened for hemoglobinopathies, enzymes and membrane defects remained without identification of the pathological mutations. This has implications at prognostic and therapeutic level, and makes the correct genetic counseling impossible.
Aims
Identify the molecular causes in a group of 14 patients with congenital HA, after screening for the most common hemoglobinopathies, enzymopathies and membranopathies.
Methods
Informed consent was signed for all the individuals in the study. We have used a targeted NGS panel encompassing the exonic regions of 24 genes (del Orbe et al, 2015) and sequenced 14 samples from patients with a congenital HA. In all the samples the common screening tests for RBC disorders were preformed, like high performance liquid chromatography (HPLC) for Hb variants, enzymatic quantification and membrane disorders screening tests as cryohemolysis test and eosin-5'-maleimide . The variants found were classified using in sillico analysis and the presence of pathological and likely pathogenic variants were confirmed by Sanger sequencing and, when available, familial studies were performed.
Results
All the samples showed a great number of variants, the majority were benign or likely benign (a media of 320/sample) or of unknown significance or likely pathogenic (media of 25/sample). Only 8 samples showed pathologic variants that can justify the clinical presentation. Most of them are associated with membrane disorders (Table). In 5 of the samples no pathological mutations were found, in one sample (sample 11) we found a NT5C3A missense mutation but in the heterozygous state (Sanger sequencing of all gene also didn’t reveal any other mutation). Three samples are compound heterozygous for mutations in the SPTA1 gene, three have a combination of mutations in different genes affecting membrane structure, one is a compound heterozygous for two missense CDAN1 mutations not described before. Sample 5 is form a sickle cell disease patient with an uncommon presentation in the newborn period, a PIEZO1 mutation inherited from the mother and a EPB41 mutation inherited from the father are likely to be the causes of the aggravated phenotype.Table – Results of the NGS sequencing – pathological variants
Conclusion
After in sillico analysis of the variants found by NGS (almost 350 per sample) and confirmation by Sanger sequencing of the pathological variants, we have identified a molecular cause for the HA in 8 of the 14 samples studied.This study confirms that target NGS is the most complete and cheaper method for sequencing the genes associated with AH but, for the cases that remain without mutations, whole exome sequencing or whole genome sequencing need to be done, in order to identify new genes that can later be added to the AH panel.
Session topic: E-poster
Keyword(s): Genotype, Hemolytic anemia
{{ help_message }}
{{filter}}