
CHANGES IN GAMMA GLOBIN MRNA LEVELS DO NOT CORRELATE WITH TOTAL HEMOGLOBIN OR FETAL HEMOGLOBIN LEVELS IN BETA THALASSEMIA INTERMEDIA PATIENTS ON HYDROXYUREA
(Abstract release date: 05/19/16)
EHA Library. Viswanathan G. 06/09/16; 133027; E1478

Dr. Ganesh Kumar Viswanathan
Contributions
Contributions
Abstract
Abstract: E1478
Type: Eposter Presentation
Background
Hydroxyurea is a hemoglobin-F inducing drug used to increase hemoglobin levels in sickle cell disease and β-thalassemia intermedia. The response to hydroxyurea is variable among individuals and its mechanism of action is less well understood. This prospective study on β-thalassemia intermedia patients aimed at analysing the effects of hydroxyurea on γ-globin mRNA levels and correlate its dynamics with changes in total hemoglobin and fetal hemoglobin levels to better understand its mechanism of action. The underlying β thalassemia mutations, α thalassemia deletions and Xmn-1Gγ (C-T) polymorphism were characterised and correlated with changes in γ-globin mRNA levels.
Aims
To study the changes in γ-globin mRNA levels in β-thalassemia intermedia patients on hydroxyurea compared to baseline levels and correlate them with changes in total hemoglobin and fetal hemoglobin levels.
Methods
Eleven newly diagnosed individuals with β-thalassemia intermedia were included in the study. Changes in γ-globin mRNA levels were compared at baseline and after 6 months of hydroxyurea therapy. These were correlated with changes in hemoglobin, hemoglobin-F, hemoglobin-A2 levels and red cell parameters. The quantitation of γ-globin mRNA levels was done by real time PCR (Roche Lightcycler 480®). ARMS-PCR was used to study the β thalassemia mutations and GAP-PCR was employed to study the α globin gene deletions. Xmn-1Gγ (C-T) polymorphism was studied by PCR-RFLP.
Results
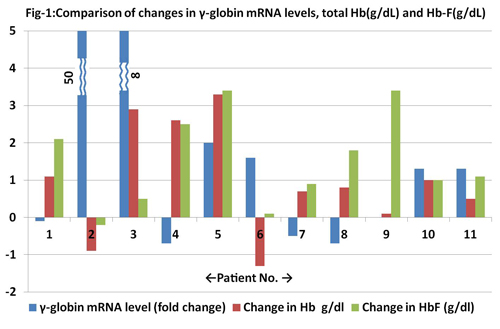
The study group included nine pediatric cases and two adult patients with age at enrolment ranging from 2 to 30 years. The male to female ratio was 1.2: 1. Four mild, five moderate and two severe β-thalassemia intermedia cases were enrolled. Milder β-thalassemia mutations were responsible in majority of cases for the thalassemia intermedia phenotype. The most frequent β-thalassemia mutation in our study group was -88 (C-T) followed by Cap+1 (A-C). All the eleven patients were negative for the 8 α-globin gene deletions tested by GAP-PCR. Seven patients were Xmn-1Gγ (-/-), two patients Xmn-1-Gγ (+/+) and two were of Xmn-1Gγ (+/-) genotype. Seven patients (63.6%) were good responders, two (18.2%) were partial and two (18.2%) were non-responders to hydroxyurea therapy. The mean increase in Hb post-HU therapy was 1 g/dl. The mean HbF increased by 1.5 g/dl in the post-HU group. Six (54.5%) patients had increased relative expression of γ-globin mRNA after HU therapy, 4 (36.4%) patients had decreased relative expression and one patient (9%) did not show any change measured by qRT-PCR. Four patients who had a decrease in relative expression of γ-globin mRNA after hydroxyurea therapy did not show a drop in Hb; rather all had a rise in Hb ranging from 0.8 to 2.6 g/dl. The relative expression of γ-globin mRNA levels did not correlate with rise in hemoglobin. Xmn-1Gγ (C-T) polymorphism was associated with high fetal hemoglobin levels at baseline and also post hydroxyurea. Hydroxyurea showed good response in majority of the patients. These results indicate that the actions of hydroxyurea might extend beyond γ-globin gene regulation. The mean hemoglobin A2 levels were decreased post hydroxyurea compared to baseline. This indicates hydroxyurea might affect regulation of other globin genes apart from the γ-globin gene. In addition there was improvement in red cell parameters (red cell indices, nucleated red cell count and reticulocyte count), bilirubin and lactate dehydrogenase levels indicating that hydroxyurea has a role in improving red cell rheology and survival.
Conclusion
In our study, the changes in relative expression of γ-globin mRNA levels (both increased and decreased levels) as measured by qRT-PCR did not directly correlate with changes in either of total hemoglobin or fetal hemoglobin levels. The increase in hemoglobin levels even in patients with decreased relative expression of γ-globin mRNA levels suggests that alternate mechanisms of action of hydroxyurea exist to increase the fetal hemoglobin and total hemoglobin in β-thalassemia intermedia patients. Xmn-1Gγ (C-T) polymorphism is associated with better response to hydroxyurea.
Session topic: E-poster
Keyword(s): Gamma globin, Hemoglobin, Hydroxyurea, Thalassemia
Type: Eposter Presentation
Background
Hydroxyurea is a hemoglobin-F inducing drug used to increase hemoglobin levels in sickle cell disease and β-thalassemia intermedia. The response to hydroxyurea is variable among individuals and its mechanism of action is less well understood. This prospective study on β-thalassemia intermedia patients aimed at analysing the effects of hydroxyurea on γ-globin mRNA levels and correlate its dynamics with changes in total hemoglobin and fetal hemoglobin levels to better understand its mechanism of action. The underlying β thalassemia mutations, α thalassemia deletions and Xmn-1Gγ (C-T) polymorphism were characterised and correlated with changes in γ-globin mRNA levels.
Aims
To study the changes in γ-globin mRNA levels in β-thalassemia intermedia patients on hydroxyurea compared to baseline levels and correlate them with changes in total hemoglobin and fetal hemoglobin levels.
Methods
Eleven newly diagnosed individuals with β-thalassemia intermedia were included in the study. Changes in γ-globin mRNA levels were compared at baseline and after 6 months of hydroxyurea therapy. These were correlated with changes in hemoglobin, hemoglobin-F, hemoglobin-A2 levels and red cell parameters. The quantitation of γ-globin mRNA levels was done by real time PCR (Roche Lightcycler 480®). ARMS-PCR was used to study the β thalassemia mutations and GAP-PCR was employed to study the α globin gene deletions. Xmn-1Gγ (C-T) polymorphism was studied by PCR-RFLP.
Results
The study group included nine pediatric cases and two adult patients with age at enrolment ranging from 2 to 30 years. The male to female ratio was 1.2: 1. Four mild, five moderate and two severe β-thalassemia intermedia cases were enrolled. Milder β-thalassemia mutations were responsible in majority of cases for the thalassemia intermedia phenotype. The most frequent β-thalassemia mutation in our study group was -88 (C-T) followed by Cap+1 (A-C). All the eleven patients were negative for the 8 α-globin gene deletions tested by GAP-PCR. Seven patients were Xmn-1Gγ (-/-), two patients Xmn-1-Gγ (+/+) and two were of Xmn-1Gγ (+/-) genotype. Seven patients (63.6%) were good responders, two (18.2%) were partial and two (18.2%) were non-responders to hydroxyurea therapy. The mean increase in Hb post-HU therapy was 1 g/dl. The mean HbF increased by 1.5 g/dl in the post-HU group. Six (54.5%) patients had increased relative expression of γ-globin mRNA after HU therapy, 4 (36.4%) patients had decreased relative expression and one patient (9%) did not show any change measured by qRT-PCR. Four patients who had a decrease in relative expression of γ-globin mRNA after hydroxyurea therapy did not show a drop in Hb; rather all had a rise in Hb ranging from 0.8 to 2.6 g/dl. The relative expression of γ-globin mRNA levels did not correlate with rise in hemoglobin. Xmn-1Gγ (C-T) polymorphism was associated with high fetal hemoglobin levels at baseline and also post hydroxyurea. Hydroxyurea showed good response in majority of the patients. These results indicate that the actions of hydroxyurea might extend beyond γ-globin gene regulation. The mean hemoglobin A2 levels were decreased post hydroxyurea compared to baseline. This indicates hydroxyurea might affect regulation of other globin genes apart from the γ-globin gene. In addition there was improvement in red cell parameters (red cell indices, nucleated red cell count and reticulocyte count), bilirubin and lactate dehydrogenase levels indicating that hydroxyurea has a role in improving red cell rheology and survival.
Conclusion
In our study, the changes in relative expression of γ-globin mRNA levels (both increased and decreased levels) as measured by qRT-PCR did not directly correlate with changes in either of total hemoglobin or fetal hemoglobin levels. The increase in hemoglobin levels even in patients with decreased relative expression of γ-globin mRNA levels suggests that alternate mechanisms of action of hydroxyurea exist to increase the fetal hemoglobin and total hemoglobin in β-thalassemia intermedia patients. Xmn-1Gγ (C-T) polymorphism is associated with better response to hydroxyurea.
Session topic: E-poster
Keyword(s): Gamma globin, Hemoglobin, Hydroxyurea, Thalassemia
Abstract: E1478
Type: Eposter Presentation
Background
Hydroxyurea is a hemoglobin-F inducing drug used to increase hemoglobin levels in sickle cell disease and β-thalassemia intermedia. The response to hydroxyurea is variable among individuals and its mechanism of action is less well understood. This prospective study on β-thalassemia intermedia patients aimed at analysing the effects of hydroxyurea on γ-globin mRNA levels and correlate its dynamics with changes in total hemoglobin and fetal hemoglobin levels to better understand its mechanism of action. The underlying β thalassemia mutations, α thalassemia deletions and Xmn-1Gγ (C-T) polymorphism were characterised and correlated with changes in γ-globin mRNA levels.
Aims
To study the changes in γ-globin mRNA levels in β-thalassemia intermedia patients on hydroxyurea compared to baseline levels and correlate them with changes in total hemoglobin and fetal hemoglobin levels.
Methods
Eleven newly diagnosed individuals with β-thalassemia intermedia were included in the study. Changes in γ-globin mRNA levels were compared at baseline and after 6 months of hydroxyurea therapy. These were correlated with changes in hemoglobin, hemoglobin-F, hemoglobin-A2 levels and red cell parameters. The quantitation of γ-globin mRNA levels was done by real time PCR (Roche Lightcycler 480®). ARMS-PCR was used to study the β thalassemia mutations and GAP-PCR was employed to study the α globin gene deletions. Xmn-1Gγ (C-T) polymorphism was studied by PCR-RFLP.
Results
The study group included nine pediatric cases and two adult patients with age at enrolment ranging from 2 to 30 years. The male to female ratio was 1.2: 1. Four mild, five moderate and two severe β-thalassemia intermedia cases were enrolled. Milder β-thalassemia mutations were responsible in majority of cases for the thalassemia intermedia phenotype. The most frequent β-thalassemia mutation in our study group was -88 (C-T) followed by Cap+1 (A-C). All the eleven patients were negative for the 8 α-globin gene deletions tested by GAP-PCR. Seven patients were Xmn-1Gγ (-/-), two patients Xmn-1-Gγ (+/+) and two were of Xmn-1Gγ (+/-) genotype. Seven patients (63.6%) were good responders, two (18.2%) were partial and two (18.2%) were non-responders to hydroxyurea therapy. The mean increase in Hb post-HU therapy was 1 g/dl. The mean HbF increased by 1.5 g/dl in the post-HU group. Six (54.5%) patients had increased relative expression of γ-globin mRNA after HU therapy, 4 (36.4%) patients had decreased relative expression and one patient (9%) did not show any change measured by qRT-PCR. Four patients who had a decrease in relative expression of γ-globin mRNA after hydroxyurea therapy did not show a drop in Hb; rather all had a rise in Hb ranging from 0.8 to 2.6 g/dl. The relative expression of γ-globin mRNA levels did not correlate with rise in hemoglobin. Xmn-1Gγ (C-T) polymorphism was associated with high fetal hemoglobin levels at baseline and also post hydroxyurea. Hydroxyurea showed good response in majority of the patients. These results indicate that the actions of hydroxyurea might extend beyond γ-globin gene regulation. The mean hemoglobin A2 levels were decreased post hydroxyurea compared to baseline. This indicates hydroxyurea might affect regulation of other globin genes apart from the γ-globin gene. In addition there was improvement in red cell parameters (red cell indices, nucleated red cell count and reticulocyte count), bilirubin and lactate dehydrogenase levels indicating that hydroxyurea has a role in improving red cell rheology and survival.
Conclusion
In our study, the changes in relative expression of γ-globin mRNA levels (both increased and decreased levels) as measured by qRT-PCR did not directly correlate with changes in either of total hemoglobin or fetal hemoglobin levels. The increase in hemoglobin levels even in patients with decreased relative expression of γ-globin mRNA levels suggests that alternate mechanisms of action of hydroxyurea exist to increase the fetal hemoglobin and total hemoglobin in β-thalassemia intermedia patients. Xmn-1Gγ (C-T) polymorphism is associated with better response to hydroxyurea.
Session topic: E-poster
Keyword(s): Gamma globin, Hemoglobin, Hydroxyurea, Thalassemia
Type: Eposter Presentation
Background
Hydroxyurea is a hemoglobin-F inducing drug used to increase hemoglobin levels in sickle cell disease and β-thalassemia intermedia. The response to hydroxyurea is variable among individuals and its mechanism of action is less well understood. This prospective study on β-thalassemia intermedia patients aimed at analysing the effects of hydroxyurea on γ-globin mRNA levels and correlate its dynamics with changes in total hemoglobin and fetal hemoglobin levels to better understand its mechanism of action. The underlying β thalassemia mutations, α thalassemia deletions and Xmn-1Gγ (C-T) polymorphism were characterised and correlated with changes in γ-globin mRNA levels.
Aims
To study the changes in γ-globin mRNA levels in β-thalassemia intermedia patients on hydroxyurea compared to baseline levels and correlate them with changes in total hemoglobin and fetal hemoglobin levels.
Methods
Eleven newly diagnosed individuals with β-thalassemia intermedia were included in the study. Changes in γ-globin mRNA levels were compared at baseline and after 6 months of hydroxyurea therapy. These were correlated with changes in hemoglobin, hemoglobin-F, hemoglobin-A2 levels and red cell parameters. The quantitation of γ-globin mRNA levels was done by real time PCR (Roche Lightcycler 480®). ARMS-PCR was used to study the β thalassemia mutations and GAP-PCR was employed to study the α globin gene deletions. Xmn-1Gγ (C-T) polymorphism was studied by PCR-RFLP.
Results
The study group included nine pediatric cases and two adult patients with age at enrolment ranging from 2 to 30 years. The male to female ratio was 1.2: 1. Four mild, five moderate and two severe β-thalassemia intermedia cases were enrolled. Milder β-thalassemia mutations were responsible in majority of cases for the thalassemia intermedia phenotype. The most frequent β-thalassemia mutation in our study group was -88 (C-T) followed by Cap+1 (A-C). All the eleven patients were negative for the 8 α-globin gene deletions tested by GAP-PCR. Seven patients were Xmn-1Gγ (-/-), two patients Xmn-1-Gγ (+/+) and two were of Xmn-1Gγ (+/-) genotype. Seven patients (63.6%) were good responders, two (18.2%) were partial and two (18.2%) were non-responders to hydroxyurea therapy. The mean increase in Hb post-HU therapy was 1 g/dl. The mean HbF increased by 1.5 g/dl in the post-HU group. Six (54.5%) patients had increased relative expression of γ-globin mRNA after HU therapy, 4 (36.4%) patients had decreased relative expression and one patient (9%) did not show any change measured by qRT-PCR. Four patients who had a decrease in relative expression of γ-globin mRNA after hydroxyurea therapy did not show a drop in Hb; rather all had a rise in Hb ranging from 0.8 to 2.6 g/dl. The relative expression of γ-globin mRNA levels did not correlate with rise in hemoglobin. Xmn-1Gγ (C-T) polymorphism was associated with high fetal hemoglobin levels at baseline and also post hydroxyurea. Hydroxyurea showed good response in majority of the patients. These results indicate that the actions of hydroxyurea might extend beyond γ-globin gene regulation. The mean hemoglobin A2 levels were decreased post hydroxyurea compared to baseline. This indicates hydroxyurea might affect regulation of other globin genes apart from the γ-globin gene. In addition there was improvement in red cell parameters (red cell indices, nucleated red cell count and reticulocyte count), bilirubin and lactate dehydrogenase levels indicating that hydroxyurea has a role in improving red cell rheology and survival.
Conclusion
In our study, the changes in relative expression of γ-globin mRNA levels (both increased and decreased levels) as measured by qRT-PCR did not directly correlate with changes in either of total hemoglobin or fetal hemoglobin levels. The increase in hemoglobin levels even in patients with decreased relative expression of γ-globin mRNA levels suggests that alternate mechanisms of action of hydroxyurea exist to increase the fetal hemoglobin and total hemoglobin in β-thalassemia intermedia patients. Xmn-1Gγ (C-T) polymorphism is associated with better response to hydroxyurea.
Session topic: E-poster
Keyword(s): Gamma globin, Hemoglobin, Hydroxyurea, Thalassemia
{{ help_message }}
{{filter}}