
NON DELETIONAL ALPHA THALASSAEMIA AND STRUCTURAL HAEMOGLOBINOPATHIES OF ALFA CHAIN MOLECULAR CHARACTERIZATION IN SPAIN IN A COHORT OF 1623 PATIENTS
(Abstract release date: 05/19/16)
EHA Library. De La Fuente Gonzalo F. 06/09/16; 133014; E1465

Dr. Felix De La Fuente Gonzalo
Contributions
Contributions
Abstract
Abstract: E1465
Type: Eposter Presentation
Background
Thalassaemias are a heterogeneous group of inherited anaemias that are characterized by the reduction or total absence of the synthesis of one or more globin chains. Structural haemoglobinopathies are inherited disorders in which the sequence of one of the globin chains, which form the haemoglobin, is altered.
Aims
To analyze the distribution of demographic variables of patients with α-thalassaemia and structural haemoglobinopathies in our region and to describe the molecular heterogeneity of both diseases and to examine the incidence of non-deletional α-thalassaemia within all cases of α-thalassaemia.
Methods
Studied Subjects: From January 2009 to December 2014, 1,623 individuals were studied from different Spanish regions (both native and immigrant populations). It was an study comprising 1,470 patients with hypochromic and microcytic anaemia, 176 subjects with a peak of abnormal haemoglobin and 23 patients who were studied for both diagnosis. Diagnostic techniques: α-thalassaemia required a conventional haemocytometer study, the reticulocyte count, the determination of Hb A2 and Hb F by ion exchange HPLC and quantification of the Hb H inclusion bodies. The diagnosis of structural haemoglobinopathies required capillary electrophoresis of haemoglobins and reversed phase or globin chain HPLC. Molecular biology techniques: automatic extraction of the DNA, discarding the most frequent large deletions and point mutations using the α-globin StripAssay®, discarding other large deletions by MLPA, and Sanger sequencing for other point mutations.
Results
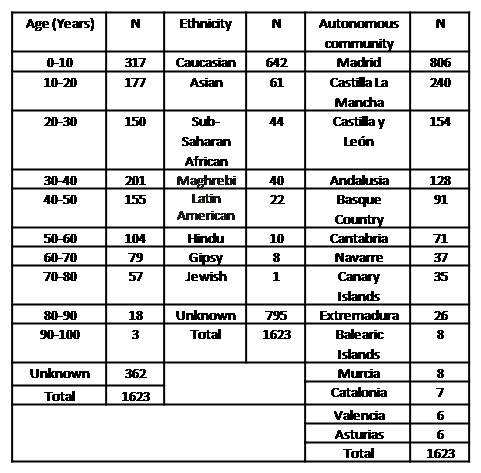
The demographics results are summarised in table 1 (Table 1: Demographics data including age, ethnicity and Autonomous community of residence of the patients). Gender balance was found in this study. Clinical diagnosis: 1 hydrops fetalis, 18 Hb H disease, 1,200 thalassaemias traits and 160 thalassaemia silent carriers were recorded within the α-thalassaemia clinical diagnostics. Regarding structural haemoglobinopathies, there were only 2 cases of haemoglobinopathies with low oxygen affinity and 1 case of haemoglobin M, which represented the only symptomatic patients. Genetic diagnosis: 1,298 carriers of the α-thalassaemia deletion were identified (905 heterozygous, 366 homozygous and 27 double heterozygous). A total of 188 patients with non-deletional α-thalassaemia were found (185 heterozygous and 3 homozygous). A total of 176 patients had structural haemoglobinopathies of the α chain (173 heterozygous and 3 homozygous). Finally, it is important to note that this study described 9 and 5 new alterations responsible for non-deletional α-thalassaemia and structural haemoglobinopathies of the α chain, respectively.
Conclusion
1)Most individuals were diagnosed with α-thalassaemia at an early age, particularly in severe cases. Further, the disease occurred predominantly in Caucasians and, to a lesser extent, in Asians. 2)Non-deletional α-thalassaemia represented 12% of all α-thalassaemias in our region, representing a higher value than described to date. 3)The most common deletion in our region was the 3.7Kb deletion that is common in Mediterranean populations, followed by Asian –SEA and --FIL. The alterations responsible for non-deletional α-thalassaemia are most represented by the Hph and Hb Groene Hart and, in the case of structural haemoglobinopathies, Hb Le Lamentin and Hb J-Paris. However, there is a high heterogeneity in the population analysed with the discovery of 36 and 35 different alterations responsible for non-deletional α-thalassaemia and structural haemoglobinopathies of the α chain, respectively.
Session topic: E-poster
Keyword(s): Genetic, Hemoglobinopathy, Thalassemia
Type: Eposter Presentation
Background
Thalassaemias are a heterogeneous group of inherited anaemias that are characterized by the reduction or total absence of the synthesis of one or more globin chains. Structural haemoglobinopathies are inherited disorders in which the sequence of one of the globin chains, which form the haemoglobin, is altered.
Aims
To analyze the distribution of demographic variables of patients with α-thalassaemia and structural haemoglobinopathies in our region and to describe the molecular heterogeneity of both diseases and to examine the incidence of non-deletional α-thalassaemia within all cases of α-thalassaemia.
Methods
Studied Subjects: From January 2009 to December 2014, 1,623 individuals were studied from different Spanish regions (both native and immigrant populations). It was an study comprising 1,470 patients with hypochromic and microcytic anaemia, 176 subjects with a peak of abnormal haemoglobin and 23 patients who were studied for both diagnosis. Diagnostic techniques: α-thalassaemia required a conventional haemocytometer study, the reticulocyte count, the determination of Hb A2 and Hb F by ion exchange HPLC and quantification of the Hb H inclusion bodies. The diagnosis of structural haemoglobinopathies required capillary electrophoresis of haemoglobins and reversed phase or globin chain HPLC. Molecular biology techniques: automatic extraction of the DNA, discarding the most frequent large deletions and point mutations using the α-globin StripAssay®, discarding other large deletions by MLPA, and Sanger sequencing for other point mutations.
Results
The demographics results are summarised in table 1 (Table 1: Demographics data including age, ethnicity and Autonomous community of residence of the patients). Gender balance was found in this study. Clinical diagnosis: 1 hydrops fetalis, 18 Hb H disease, 1,200 thalassaemias traits and 160 thalassaemia silent carriers were recorded within the α-thalassaemia clinical diagnostics. Regarding structural haemoglobinopathies, there were only 2 cases of haemoglobinopathies with low oxygen affinity and 1 case of haemoglobin M, which represented the only symptomatic patients. Genetic diagnosis: 1,298 carriers of the α-thalassaemia deletion were identified (905 heterozygous, 366 homozygous and 27 double heterozygous). A total of 188 patients with non-deletional α-thalassaemia were found (185 heterozygous and 3 homozygous). A total of 176 patients had structural haemoglobinopathies of the α chain (173 heterozygous and 3 homozygous). Finally, it is important to note that this study described 9 and 5 new alterations responsible for non-deletional α-thalassaemia and structural haemoglobinopathies of the α chain, respectively.
Conclusion
1)Most individuals were diagnosed with α-thalassaemia at an early age, particularly in severe cases. Further, the disease occurred predominantly in Caucasians and, to a lesser extent, in Asians. 2)Non-deletional α-thalassaemia represented 12% of all α-thalassaemias in our region, representing a higher value than described to date. 3)The most common deletion in our region was the 3.7Kb deletion that is common in Mediterranean populations, followed by Asian –SEA and --FIL. The alterations responsible for non-deletional α-thalassaemia are most represented by the Hph and Hb Groene Hart and, in the case of structural haemoglobinopathies, Hb Le Lamentin and Hb J-Paris. However, there is a high heterogeneity in the population analysed with the discovery of 36 and 35 different alterations responsible for non-deletional α-thalassaemia and structural haemoglobinopathies of the α chain, respectively.
Session topic: E-poster
Keyword(s): Genetic, Hemoglobinopathy, Thalassemia
Abstract: E1465
Type: Eposter Presentation
Background
Thalassaemias are a heterogeneous group of inherited anaemias that are characterized by the reduction or total absence of the synthesis of one or more globin chains. Structural haemoglobinopathies are inherited disorders in which the sequence of one of the globin chains, which form the haemoglobin, is altered.
Aims
To analyze the distribution of demographic variables of patients with α-thalassaemia and structural haemoglobinopathies in our region and to describe the molecular heterogeneity of both diseases and to examine the incidence of non-deletional α-thalassaemia within all cases of α-thalassaemia.
Methods
Studied Subjects: From January 2009 to December 2014, 1,623 individuals were studied from different Spanish regions (both native and immigrant populations). It was an study comprising 1,470 patients with hypochromic and microcytic anaemia, 176 subjects with a peak of abnormal haemoglobin and 23 patients who were studied for both diagnosis. Diagnostic techniques: α-thalassaemia required a conventional haemocytometer study, the reticulocyte count, the determination of Hb A2 and Hb F by ion exchange HPLC and quantification of the Hb H inclusion bodies. The diagnosis of structural haemoglobinopathies required capillary electrophoresis of haemoglobins and reversed phase or globin chain HPLC. Molecular biology techniques: automatic extraction of the DNA, discarding the most frequent large deletions and point mutations using the α-globin StripAssay®, discarding other large deletions by MLPA, and Sanger sequencing for other point mutations.
Results
The demographics results are summarised in table 1 (Table 1: Demographics data including age, ethnicity and Autonomous community of residence of the patients). Gender balance was found in this study. Clinical diagnosis: 1 hydrops fetalis, 18 Hb H disease, 1,200 thalassaemias traits and 160 thalassaemia silent carriers were recorded within the α-thalassaemia clinical diagnostics. Regarding structural haemoglobinopathies, there were only 2 cases of haemoglobinopathies with low oxygen affinity and 1 case of haemoglobin M, which represented the only symptomatic patients. Genetic diagnosis: 1,298 carriers of the α-thalassaemia deletion were identified (905 heterozygous, 366 homozygous and 27 double heterozygous). A total of 188 patients with non-deletional α-thalassaemia were found (185 heterozygous and 3 homozygous). A total of 176 patients had structural haemoglobinopathies of the α chain (173 heterozygous and 3 homozygous). Finally, it is important to note that this study described 9 and 5 new alterations responsible for non-deletional α-thalassaemia and structural haemoglobinopathies of the α chain, respectively.
Conclusion
1)Most individuals were diagnosed with α-thalassaemia at an early age, particularly in severe cases. Further, the disease occurred predominantly in Caucasians and, to a lesser extent, in Asians. 2)Non-deletional α-thalassaemia represented 12% of all α-thalassaemias in our region, representing a higher value than described to date. 3)The most common deletion in our region was the 3.7Kb deletion that is common in Mediterranean populations, followed by Asian –SEA and --FIL. The alterations responsible for non-deletional α-thalassaemia are most represented by the Hph and Hb Groene Hart and, in the case of structural haemoglobinopathies, Hb Le Lamentin and Hb J-Paris. However, there is a high heterogeneity in the population analysed with the discovery of 36 and 35 different alterations responsible for non-deletional α-thalassaemia and structural haemoglobinopathies of the α chain, respectively.
Session topic: E-poster
Keyword(s): Genetic, Hemoglobinopathy, Thalassemia
Type: Eposter Presentation
Background
Thalassaemias are a heterogeneous group of inherited anaemias that are characterized by the reduction or total absence of the synthesis of one or more globin chains. Structural haemoglobinopathies are inherited disorders in which the sequence of one of the globin chains, which form the haemoglobin, is altered.
Aims
To analyze the distribution of demographic variables of patients with α-thalassaemia and structural haemoglobinopathies in our region and to describe the molecular heterogeneity of both diseases and to examine the incidence of non-deletional α-thalassaemia within all cases of α-thalassaemia.
Methods
Studied Subjects: From January 2009 to December 2014, 1,623 individuals were studied from different Spanish regions (both native and immigrant populations). It was an study comprising 1,470 patients with hypochromic and microcytic anaemia, 176 subjects with a peak of abnormal haemoglobin and 23 patients who were studied for both diagnosis. Diagnostic techniques: α-thalassaemia required a conventional haemocytometer study, the reticulocyte count, the determination of Hb A2 and Hb F by ion exchange HPLC and quantification of the Hb H inclusion bodies. The diagnosis of structural haemoglobinopathies required capillary electrophoresis of haemoglobins and reversed phase or globin chain HPLC. Molecular biology techniques: automatic extraction of the DNA, discarding the most frequent large deletions and point mutations using the α-globin StripAssay®, discarding other large deletions by MLPA, and Sanger sequencing for other point mutations.
Results
The demographics results are summarised in table 1 (Table 1: Demographics data including age, ethnicity and Autonomous community of residence of the patients). Gender balance was found in this study. Clinical diagnosis: 1 hydrops fetalis, 18 Hb H disease, 1,200 thalassaemias traits and 160 thalassaemia silent carriers were recorded within the α-thalassaemia clinical diagnostics. Regarding structural haemoglobinopathies, there were only 2 cases of haemoglobinopathies with low oxygen affinity and 1 case of haemoglobin M, which represented the only symptomatic patients. Genetic diagnosis: 1,298 carriers of the α-thalassaemia deletion were identified (905 heterozygous, 366 homozygous and 27 double heterozygous). A total of 188 patients with non-deletional α-thalassaemia were found (185 heterozygous and 3 homozygous). A total of 176 patients had structural haemoglobinopathies of the α chain (173 heterozygous and 3 homozygous). Finally, it is important to note that this study described 9 and 5 new alterations responsible for non-deletional α-thalassaemia and structural haemoglobinopathies of the α chain, respectively.
Conclusion
1)Most individuals were diagnosed with α-thalassaemia at an early age, particularly in severe cases. Further, the disease occurred predominantly in Caucasians and, to a lesser extent, in Asians. 2)Non-deletional α-thalassaemia represented 12% of all α-thalassaemias in our region, representing a higher value than described to date. 3)The most common deletion in our region was the 3.7Kb deletion that is common in Mediterranean populations, followed by Asian –SEA and --FIL. The alterations responsible for non-deletional α-thalassaemia are most represented by the Hph and Hb Groene Hart and, in the case of structural haemoglobinopathies, Hb Le Lamentin and Hb J-Paris. However, there is a high heterogeneity in the population analysed with the discovery of 36 and 35 different alterations responsible for non-deletional α-thalassaemia and structural haemoglobinopathies of the α chain, respectively.
Session topic: E-poster
Keyword(s): Genetic, Hemoglobinopathy, Thalassemia
{{ help_message }}
{{filter}}