
FAMILIAL HEMOPHAGOCYTIC LYMPHOHISTIOCYTOSIS IN OMAN: AN UPDATE ON UNIQUE CLINICAL AND MOLECULAR FEATURES
(Abstract release date: 05/19/16)
EHA Library. Elshinawy M. 06/09/16; 132958; E1409
Disclosure(s): Nothing to disclose

Assoc. Prof. Mohamed Elshinawy
Contributions
Contributions
Abstract
Abstract: E1409
Type: Eposter Presentation
Background
Familial hemophagocytic lymphohistiocytosis (FHLH) is a rare life-threatening autosomal recessive disorder. Due to high rate of consanguineous marriage in Oman, the incidence of HLH is relatively higher in this part of the world. Clinically, HLH is characterized by unexplained high-grade fever and splenomegaly associated with bi/pancytopenia, hyperferrtinemia, hypofibrinogenemia and hypertriglyceridemia.
Aims
To review the clinical features, laboratory findings, molecular characteristics, course and outcome of all cases of FHLH in Oman.
Methods
A retrospective/prospective data analysis of all patients diagnosed with FHLH in Oman during the period from January 1997 to February 2016 was done. Flowcytometry has been used as a fast method to detect perforin expression at diagnosis. Direct DNA sequencing was done to specify mutations in patients and their families. Our results have been initially confirmed and validated by Karolinska Institute, Sweden.
Results
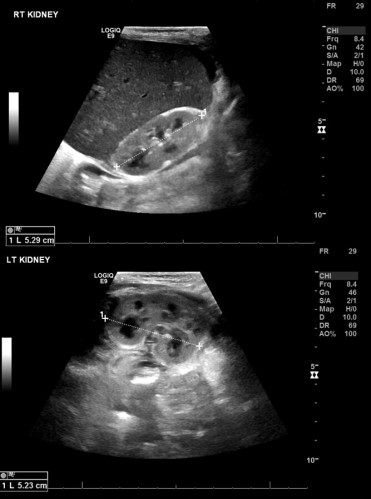
Forty-eight patients (26 males & 22 females) with FHLH were identified. Their median age was 88 days with a range of (3 days– 13 months). Thirty-seven patients (77%) had positive consanguinity, while 25 (52%) had positive family history. Forty-one patients (85.4%) presented with fever and 44 (91.6%) had splenomegaly. Twenty-seven cases (56.25 %) had evidence of central nervous system involvement detected either at diagnosis or during the course of therapy. Atypical rare clinical presentations included capillary leak syndrome in 5 cases (10.5%), severe hypertrophic obstructive cardiomyopathy in 2 siblings after receiving high dose dexamethasone, and bilateral renal infiltration as a unique clinical feature in a 2 month-old male infant (Figure 1). Of note, renal involvement in this patient was completely reversed after 4 weeks of chemotherapy.Bicytopenia was found in 39 (81.3%) infants, hypertriglyceridemia in 28 (58.3%) and hypofibrinogenemia in 45 (93.8%). All patients had serum ferritin>500 ng/ml.At molecular level, the most commonly detected genetic abnormality was perforin gene mutation (FHLH2), identified in 14 out of 17 studied families (82.4%). Syntaxin 11 mutation (FHL4) was detected in only one patient, while MUNC 13-4 mutation was not found. Two families did not show any mutations. The high incidence of perforin gene mutations may denote a common genetic pool of FHLH cases in Oman.Twenty-three (47.9 %) cases passed away. Fifteen infants were referred late after being treated with antibiotics in peripheral hospitals as presumed cases of sepsis. Our standard of care is HLH-2004 protocol, followed by hematopoietic stem cell transplant (HSCT). Twenty-one patients were successfully transplanted in our center. It is noteworthy that three of them have received haploidentical HSCT.
Conclusion
We report an updated registry of FHLH cases in Oman, including typical and atypical unique clinical presentations, characteristic molecular features, course of the disease and treatment outcome. Taken into consideration the relatively higher incidence of FHLH in this part of the world, we recommend premarital screening, implementation of pre-implantation genetic diagnosis (PGD) for high risk families and raising the awareness of health care professionals in order to suspect and refer new cases as early as possible.
Session topic: E-poster
Type: Eposter Presentation
Background
Familial hemophagocytic lymphohistiocytosis (FHLH) is a rare life-threatening autosomal recessive disorder. Due to high rate of consanguineous marriage in Oman, the incidence of HLH is relatively higher in this part of the world. Clinically, HLH is characterized by unexplained high-grade fever and splenomegaly associated with bi/pancytopenia, hyperferrtinemia, hypofibrinogenemia and hypertriglyceridemia.
Aims
To review the clinical features, laboratory findings, molecular characteristics, course and outcome of all cases of FHLH in Oman.
Methods
A retrospective/prospective data analysis of all patients diagnosed with FHLH in Oman during the period from January 1997 to February 2016 was done. Flowcytometry has been used as a fast method to detect perforin expression at diagnosis. Direct DNA sequencing was done to specify mutations in patients and their families. Our results have been initially confirmed and validated by Karolinska Institute, Sweden.
Results
Forty-eight patients (26 males & 22 females) with FHLH were identified. Their median age was 88 days with a range of (3 days– 13 months). Thirty-seven patients (77%) had positive consanguinity, while 25 (52%) had positive family history. Forty-one patients (85.4%) presented with fever and 44 (91.6%) had splenomegaly. Twenty-seven cases (56.25 %) had evidence of central nervous system involvement detected either at diagnosis or during the course of therapy. Atypical rare clinical presentations included capillary leak syndrome in 5 cases (10.5%), severe hypertrophic obstructive cardiomyopathy in 2 siblings after receiving high dose dexamethasone, and bilateral renal infiltration as a unique clinical feature in a 2 month-old male infant (Figure 1). Of note, renal involvement in this patient was completely reversed after 4 weeks of chemotherapy.Bicytopenia was found in 39 (81.3%) infants, hypertriglyceridemia in 28 (58.3%) and hypofibrinogenemia in 45 (93.8%). All patients had serum ferritin>500 ng/ml.At molecular level, the most commonly detected genetic abnormality was perforin gene mutation (FHLH2), identified in 14 out of 17 studied families (82.4%). Syntaxin 11 mutation (FHL4) was detected in only one patient, while MUNC 13-4 mutation was not found. Two families did not show any mutations. The high incidence of perforin gene mutations may denote a common genetic pool of FHLH cases in Oman.Twenty-three (47.9 %) cases passed away. Fifteen infants were referred late after being treated with antibiotics in peripheral hospitals as presumed cases of sepsis. Our standard of care is HLH-2004 protocol, followed by hematopoietic stem cell transplant (HSCT). Twenty-one patients were successfully transplanted in our center. It is noteworthy that three of them have received haploidentical HSCT.
Conclusion
We report an updated registry of FHLH cases in Oman, including typical and atypical unique clinical presentations, characteristic molecular features, course of the disease and treatment outcome. Taken into consideration the relatively higher incidence of FHLH in this part of the world, we recommend premarital screening, implementation of pre-implantation genetic diagnosis (PGD) for high risk families and raising the awareness of health care professionals in order to suspect and refer new cases as early as possible.
Session topic: E-poster
Abstract: E1409
Type: Eposter Presentation
Background
Familial hemophagocytic lymphohistiocytosis (FHLH) is a rare life-threatening autosomal recessive disorder. Due to high rate of consanguineous marriage in Oman, the incidence of HLH is relatively higher in this part of the world. Clinically, HLH is characterized by unexplained high-grade fever and splenomegaly associated with bi/pancytopenia, hyperferrtinemia, hypofibrinogenemia and hypertriglyceridemia.
Aims
To review the clinical features, laboratory findings, molecular characteristics, course and outcome of all cases of FHLH in Oman.
Methods
A retrospective/prospective data analysis of all patients diagnosed with FHLH in Oman during the period from January 1997 to February 2016 was done. Flowcytometry has been used as a fast method to detect perforin expression at diagnosis. Direct DNA sequencing was done to specify mutations in patients and their families. Our results have been initially confirmed and validated by Karolinska Institute, Sweden.
Results
Forty-eight patients (26 males & 22 females) with FHLH were identified. Their median age was 88 days with a range of (3 days– 13 months). Thirty-seven patients (77%) had positive consanguinity, while 25 (52%) had positive family history. Forty-one patients (85.4%) presented with fever and 44 (91.6%) had splenomegaly. Twenty-seven cases (56.25 %) had evidence of central nervous system involvement detected either at diagnosis or during the course of therapy. Atypical rare clinical presentations included capillary leak syndrome in 5 cases (10.5%), severe hypertrophic obstructive cardiomyopathy in 2 siblings after receiving high dose dexamethasone, and bilateral renal infiltration as a unique clinical feature in a 2 month-old male infant (Figure 1). Of note, renal involvement in this patient was completely reversed after 4 weeks of chemotherapy.Bicytopenia was found in 39 (81.3%) infants, hypertriglyceridemia in 28 (58.3%) and hypofibrinogenemia in 45 (93.8%). All patients had serum ferritin>500 ng/ml.At molecular level, the most commonly detected genetic abnormality was perforin gene mutation (FHLH2), identified in 14 out of 17 studied families (82.4%). Syntaxin 11 mutation (FHL4) was detected in only one patient, while MUNC 13-4 mutation was not found. Two families did not show any mutations. The high incidence of perforin gene mutations may denote a common genetic pool of FHLH cases in Oman.Twenty-three (47.9 %) cases passed away. Fifteen infants were referred late after being treated with antibiotics in peripheral hospitals as presumed cases of sepsis. Our standard of care is HLH-2004 protocol, followed by hematopoietic stem cell transplant (HSCT). Twenty-one patients were successfully transplanted in our center. It is noteworthy that three of them have received haploidentical HSCT.
Conclusion
We report an updated registry of FHLH cases in Oman, including typical and atypical unique clinical presentations, characteristic molecular features, course of the disease and treatment outcome. Taken into consideration the relatively higher incidence of FHLH in this part of the world, we recommend premarital screening, implementation of pre-implantation genetic diagnosis (PGD) for high risk families and raising the awareness of health care professionals in order to suspect and refer new cases as early as possible.
Session topic: E-poster
Type: Eposter Presentation
Background
Familial hemophagocytic lymphohistiocytosis (FHLH) is a rare life-threatening autosomal recessive disorder. Due to high rate of consanguineous marriage in Oman, the incidence of HLH is relatively higher in this part of the world. Clinically, HLH is characterized by unexplained high-grade fever and splenomegaly associated with bi/pancytopenia, hyperferrtinemia, hypofibrinogenemia and hypertriglyceridemia.
Aims
To review the clinical features, laboratory findings, molecular characteristics, course and outcome of all cases of FHLH in Oman.
Methods
A retrospective/prospective data analysis of all patients diagnosed with FHLH in Oman during the period from January 1997 to February 2016 was done. Flowcytometry has been used as a fast method to detect perforin expression at diagnosis. Direct DNA sequencing was done to specify mutations in patients and their families. Our results have been initially confirmed and validated by Karolinska Institute, Sweden.
Results
Forty-eight patients (26 males & 22 females) with FHLH were identified. Their median age was 88 days with a range of (3 days– 13 months). Thirty-seven patients (77%) had positive consanguinity, while 25 (52%) had positive family history. Forty-one patients (85.4%) presented with fever and 44 (91.6%) had splenomegaly. Twenty-seven cases (56.25 %) had evidence of central nervous system involvement detected either at diagnosis or during the course of therapy. Atypical rare clinical presentations included capillary leak syndrome in 5 cases (10.5%), severe hypertrophic obstructive cardiomyopathy in 2 siblings after receiving high dose dexamethasone, and bilateral renal infiltration as a unique clinical feature in a 2 month-old male infant (Figure 1). Of note, renal involvement in this patient was completely reversed after 4 weeks of chemotherapy.Bicytopenia was found in 39 (81.3%) infants, hypertriglyceridemia in 28 (58.3%) and hypofibrinogenemia in 45 (93.8%). All patients had serum ferritin>500 ng/ml.At molecular level, the most commonly detected genetic abnormality was perforin gene mutation (FHLH2), identified in 14 out of 17 studied families (82.4%). Syntaxin 11 mutation (FHL4) was detected in only one patient, while MUNC 13-4 mutation was not found. Two families did not show any mutations. The high incidence of perforin gene mutations may denote a common genetic pool of FHLH cases in Oman.Twenty-three (47.9 %) cases passed away. Fifteen infants were referred late after being treated with antibiotics in peripheral hospitals as presumed cases of sepsis. Our standard of care is HLH-2004 protocol, followed by hematopoietic stem cell transplant (HSCT). Twenty-one patients were successfully transplanted in our center. It is noteworthy that three of them have received haploidentical HSCT.
Conclusion
We report an updated registry of FHLH cases in Oman, including typical and atypical unique clinical presentations, characteristic molecular features, course of the disease and treatment outcome. Taken into consideration the relatively higher incidence of FHLH in this part of the world, we recommend premarital screening, implementation of pre-implantation genetic diagnosis (PGD) for high risk families and raising the awareness of health care professionals in order to suspect and refer new cases as early as possible.
Session topic: E-poster
{{ help_message }}
{{filter}}