
PROPOSAL FOR A REVISED PROGNOSTIC MODEL IN PRIMARY MYELOFIBROSIS THAT ACCOUNTS FOR JAK2, MPL AND CALR MUTATIONS :AN EXPERIENCE OF A SINGLE CENTER IN CHINA
(Abstract release date: 05/19/16)
EHA Library. Li B. 06/09/16; 132903; E1354

Dr. Bing Li
Contributions
Contributions
Abstract
Abstract: E1354
Type: Eposter Presentation
Background
Primary myelofibrosis (PMF) is a Philadelphia-chromosome-negative myeloproliferative neoplasm (MPN) in which JAK2V617F, CALR and MPLW515 mutations are the most frequent driver mutations. PMF appears to have different clinical features in different population. For example, Chinese patients have different clinical and laboratory features compared with PMF in patients of predominately European descent. These differences may affect prognostic scoring systems which might require revision based on the population being evaluated.
Aims
In current study, we focus on the contribution of driver mutations (JAK2, MPL and CALR) to survival and propose a clinical-molecular prognostic model based on the data from a single center in China.
Methods
402 consecutive subjects with PMF had a bone marrow sample collected at diagnosis or referral, February,1990 to February, 2015 at the Institute of Hematology and Blood Disease Hospital, Chinese Academy of Medical Sciences were enrolled. Follow-up data were available for 361 subjects. Median follow-up of survivors was 36 months (range, 1-385) months. Patients were divided randomly into a study group (n=279) and a test group (n=123). The impact of JAK2, MPL or CALR mutations on prognosis was analyzed by univariate- and multivariate survival analyses.
Results
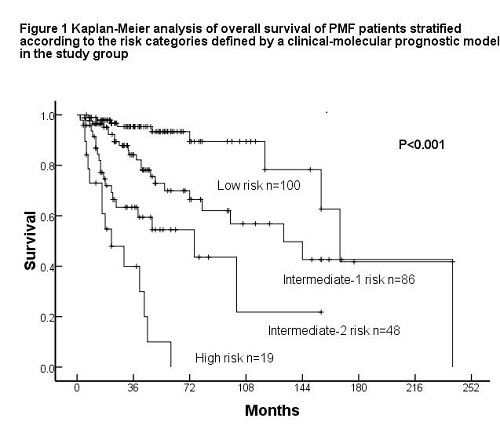
JAK2V617F mutations were detected in 189 subjects (47%), MPLW515 mutations in 13 (3.2%) and CALR mutations in 81 (20.1%). 119 subjects (29.6%) had no detectable mutation in JAK2, MPL or CALR. There were 30 (37%) type-1 and 48 (59.3%) type-2 CALR mutations. In the study group, patients with CALR type-2 mutations or no detectable mutation had briefer survival compared to those with JAK2, MPL or CALR type-1 or other less common CALR mutations (HR 2.99, 95% CI 1.935-4.619, P<0.001). Therefore, patients were categorized into the high-risk with CALR type-2 mutations or no detectable driver mutation and the low-risk without aforementioned mutations status. A multivariate analysis of prognostic factors in the study group identified the following independent factors (P<0.05): Dynamic International Prognostic Scoring System (DIPSS) risk group, palpable spleen and the status of JAK2, MPL or CALR mutations. We assigned each factor a weight: (1) 3 for DIPSS high-risk; (2) 2 for DIPSS intermediate-2 risk; (3) 1 for DIPSS intermediate-1 risk; (4) 0.5 for no splenomegaly, (5) 1 for CALR type-2 mutation or non-mutated JAK2, MPL and CALR. Subjects were categorized into 4 risk cohorts: (1) low (score 0-1; 39.5% of patients); (2) intermediate-1 (score 1.5-2; 34% of patients); intermediate-2 (score 2.5-3; 19% of patients); and high (score ≥3.5; 7.5% of patients; Fig. 2A).The revised clinical-molecular prognostic model divided patients into 4 prognostic groups with significantly different outcomes (Figure 1) and had a significantly higher predictive power for survival than the DIPSS. The model was validated in the test group.
Conclusion
This study indicates that the prognostic system including driver mutations (JAK2, MPL and CALR) status could more accurately evaluate survival for patients with PMF.
Session topic: E-poster
Keyword(s): Mutation analysis, Myelofibrosis, Prognosis
Type: Eposter Presentation
Background
Primary myelofibrosis (PMF) is a Philadelphia-chromosome-negative myeloproliferative neoplasm (MPN) in which JAK2V617F, CALR and MPLW515 mutations are the most frequent driver mutations. PMF appears to have different clinical features in different population. For example, Chinese patients have different clinical and laboratory features compared with PMF in patients of predominately European descent. These differences may affect prognostic scoring systems which might require revision based on the population being evaluated.
Aims
In current study, we focus on the contribution of driver mutations (JAK2, MPL and CALR) to survival and propose a clinical-molecular prognostic model based on the data from a single center in China.
Methods
402 consecutive subjects with PMF had a bone marrow sample collected at diagnosis or referral, February,1990 to February, 2015 at the Institute of Hematology and Blood Disease Hospital, Chinese Academy of Medical Sciences were enrolled. Follow-up data were available for 361 subjects. Median follow-up of survivors was 36 months (range, 1-385) months. Patients were divided randomly into a study group (n=279) and a test group (n=123). The impact of JAK2, MPL or CALR mutations on prognosis was analyzed by univariate- and multivariate survival analyses.
Results
JAK2V617F mutations were detected in 189 subjects (47%), MPLW515 mutations in 13 (3.2%) and CALR mutations in 81 (20.1%). 119 subjects (29.6%) had no detectable mutation in JAK2, MPL or CALR. There were 30 (37%) type-1 and 48 (59.3%) type-2 CALR mutations. In the study group, patients with CALR type-2 mutations or no detectable mutation had briefer survival compared to those with JAK2, MPL or CALR type-1 or other less common CALR mutations (HR 2.99, 95% CI 1.935-4.619, P<0.001). Therefore, patients were categorized into the high-risk with CALR type-2 mutations or no detectable driver mutation and the low-risk without aforementioned mutations status. A multivariate analysis of prognostic factors in the study group identified the following independent factors (P<0.05): Dynamic International Prognostic Scoring System (DIPSS) risk group, palpable spleen and the status of JAK2, MPL or CALR mutations. We assigned each factor a weight: (1) 3 for DIPSS high-risk; (2) 2 for DIPSS intermediate-2 risk; (3) 1 for DIPSS intermediate-1 risk; (4) 0.5 for no splenomegaly, (5) 1 for CALR type-2 mutation or non-mutated JAK2, MPL and CALR. Subjects were categorized into 4 risk cohorts: (1) low (score 0-1; 39.5% of patients); (2) intermediate-1 (score 1.5-2; 34% of patients); intermediate-2 (score 2.5-3; 19% of patients); and high (score ≥3.5; 7.5% of patients; Fig. 2A).The revised clinical-molecular prognostic model divided patients into 4 prognostic groups with significantly different outcomes (Figure 1) and had a significantly higher predictive power for survival than the DIPSS. The model was validated in the test group.
Conclusion
This study indicates that the prognostic system including driver mutations (JAK2, MPL and CALR) status could more accurately evaluate survival for patients with PMF.
Session topic: E-poster
Keyword(s): Mutation analysis, Myelofibrosis, Prognosis
Abstract: E1354
Type: Eposter Presentation
Background
Primary myelofibrosis (PMF) is a Philadelphia-chromosome-negative myeloproliferative neoplasm (MPN) in which JAK2V617F, CALR and MPLW515 mutations are the most frequent driver mutations. PMF appears to have different clinical features in different population. For example, Chinese patients have different clinical and laboratory features compared with PMF in patients of predominately European descent. These differences may affect prognostic scoring systems which might require revision based on the population being evaluated.
Aims
In current study, we focus on the contribution of driver mutations (JAK2, MPL and CALR) to survival and propose a clinical-molecular prognostic model based on the data from a single center in China.
Methods
402 consecutive subjects with PMF had a bone marrow sample collected at diagnosis or referral, February,1990 to February, 2015 at the Institute of Hematology and Blood Disease Hospital, Chinese Academy of Medical Sciences were enrolled. Follow-up data were available for 361 subjects. Median follow-up of survivors was 36 months (range, 1-385) months. Patients were divided randomly into a study group (n=279) and a test group (n=123). The impact of JAK2, MPL or CALR mutations on prognosis was analyzed by univariate- and multivariate survival analyses.
Results
JAK2V617F mutations were detected in 189 subjects (47%), MPLW515 mutations in 13 (3.2%) and CALR mutations in 81 (20.1%). 119 subjects (29.6%) had no detectable mutation in JAK2, MPL or CALR. There were 30 (37%) type-1 and 48 (59.3%) type-2 CALR mutations. In the study group, patients with CALR type-2 mutations or no detectable mutation had briefer survival compared to those with JAK2, MPL or CALR type-1 or other less common CALR mutations (HR 2.99, 95% CI 1.935-4.619, P<0.001). Therefore, patients were categorized into the high-risk with CALR type-2 mutations or no detectable driver mutation and the low-risk without aforementioned mutations status. A multivariate analysis of prognostic factors in the study group identified the following independent factors (P<0.05): Dynamic International Prognostic Scoring System (DIPSS) risk group, palpable spleen and the status of JAK2, MPL or CALR mutations. We assigned each factor a weight: (1) 3 for DIPSS high-risk; (2) 2 for DIPSS intermediate-2 risk; (3) 1 for DIPSS intermediate-1 risk; (4) 0.5 for no splenomegaly, (5) 1 for CALR type-2 mutation or non-mutated JAK2, MPL and CALR. Subjects were categorized into 4 risk cohorts: (1) low (score 0-1; 39.5% of patients); (2) intermediate-1 (score 1.5-2; 34% of patients); intermediate-2 (score 2.5-3; 19% of patients); and high (score ≥3.5; 7.5% of patients; Fig. 2A).The revised clinical-molecular prognostic model divided patients into 4 prognostic groups with significantly different outcomes (Figure 1) and had a significantly higher predictive power for survival than the DIPSS. The model was validated in the test group.
Conclusion
This study indicates that the prognostic system including driver mutations (JAK2, MPL and CALR) status could more accurately evaluate survival for patients with PMF.
Session topic: E-poster
Keyword(s): Mutation analysis, Myelofibrosis, Prognosis
Type: Eposter Presentation
Background
Primary myelofibrosis (PMF) is a Philadelphia-chromosome-negative myeloproliferative neoplasm (MPN) in which JAK2V617F, CALR and MPLW515 mutations are the most frequent driver mutations. PMF appears to have different clinical features in different population. For example, Chinese patients have different clinical and laboratory features compared with PMF in patients of predominately European descent. These differences may affect prognostic scoring systems which might require revision based on the population being evaluated.
Aims
In current study, we focus on the contribution of driver mutations (JAK2, MPL and CALR) to survival and propose a clinical-molecular prognostic model based on the data from a single center in China.
Methods
402 consecutive subjects with PMF had a bone marrow sample collected at diagnosis or referral, February,1990 to February, 2015 at the Institute of Hematology and Blood Disease Hospital, Chinese Academy of Medical Sciences were enrolled. Follow-up data were available for 361 subjects. Median follow-up of survivors was 36 months (range, 1-385) months. Patients were divided randomly into a study group (n=279) and a test group (n=123). The impact of JAK2, MPL or CALR mutations on prognosis was analyzed by univariate- and multivariate survival analyses.
Results
JAK2V617F mutations were detected in 189 subjects (47%), MPLW515 mutations in 13 (3.2%) and CALR mutations in 81 (20.1%). 119 subjects (29.6%) had no detectable mutation in JAK2, MPL or CALR. There were 30 (37%) type-1 and 48 (59.3%) type-2 CALR mutations. In the study group, patients with CALR type-2 mutations or no detectable mutation had briefer survival compared to those with JAK2, MPL or CALR type-1 or other less common CALR mutations (HR 2.99, 95% CI 1.935-4.619, P<0.001). Therefore, patients were categorized into the high-risk with CALR type-2 mutations or no detectable driver mutation and the low-risk without aforementioned mutations status. A multivariate analysis of prognostic factors in the study group identified the following independent factors (P<0.05): Dynamic International Prognostic Scoring System (DIPSS) risk group, palpable spleen and the status of JAK2, MPL or CALR mutations. We assigned each factor a weight: (1) 3 for DIPSS high-risk; (2) 2 for DIPSS intermediate-2 risk; (3) 1 for DIPSS intermediate-1 risk; (4) 0.5 for no splenomegaly, (5) 1 for CALR type-2 mutation or non-mutated JAK2, MPL and CALR. Subjects were categorized into 4 risk cohorts: (1) low (score 0-1; 39.5% of patients); (2) intermediate-1 (score 1.5-2; 34% of patients); intermediate-2 (score 2.5-3; 19% of patients); and high (score ≥3.5; 7.5% of patients; Fig. 2A).The revised clinical-molecular prognostic model divided patients into 4 prognostic groups with significantly different outcomes (Figure 1) and had a significantly higher predictive power for survival than the DIPSS. The model was validated in the test group.
Conclusion
This study indicates that the prognostic system including driver mutations (JAK2, MPL and CALR) status could more accurately evaluate survival for patients with PMF.
Session topic: E-poster
Keyword(s): Mutation analysis, Myelofibrosis, Prognosis
{{ help_message }}
{{filter}}