
CLONAL DYNAMICS OF TWO DISTINCT CLONES IN LEUKEMIC TRANSFORMATION FROM MDS WITH ISOLATED DEL(5Q) HARBORING TP53 MUTATION
(Abstract release date: 05/19/16)
EHA Library. Tsukamoto H. 06/09/16; 132753; E1204

Dr. Hiroyuki Tsukamoto
Contributions
Contributions
Abstract
Abstract: E1204
Type: Eposter Presentation
Background
Although myelodysplastic syndromes (MDS) with isolated del(5q) represents a favorable prognosis, the presence of TP53 mutation has been associated with transformation to secondary AML (sAML) accompanied by the emergence of a complex karyotype (CK). The demethylating agent azacitidine (AZA) is currently the standard therapy for patients with high-risk MDS. However, some reports showed AZA treatment cannot improve survival in high-risk MDS and sAML patients with TP53 mutations, in spite of response to AZA. The molecular mechanisms in the acquisition of resistance to AZA in TP53 mutated cases has not been completely understood.
Aims
To clarify the molecular mechanisms underlying disease progression and the acquisition of chemo-resistance in MDS, we delineated clonal evolution during disease course using high throughput sequencing of serial samples.
Methods
The 77-year-old female patient was diagnosed with MDS with isolated del(5q). She then received lenalidomide (LEN), but progressed to sAML with CK involving -7. She received AZA with LEN and then achieved temporarily marrow CR (blast<5%). Although she received same therapies, she relapsed and died of progressive leukemia. Genomic DNA was collected at four stages (RAEB-1, sAML, remission, and relapse) on informed consent. We performed whole exome sequencing (WES) and PCR amplicon based target deep sequencing by NGS (Illumina HiSeq 2500). Copy number alterations (CNA) were analyzed based on the read number and allelic imbalances were estimated from allele frequency of heterozygous SNPs.
Results
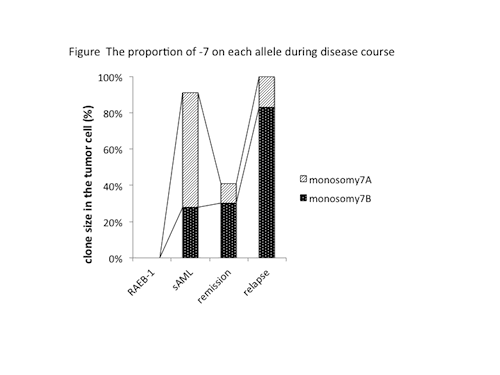
The mean coverage of WES and target deep sequencing was 209 times and 67073 times, respectively. We detected 15 somatic single nucleotide variants (SNV) and 2 frameshift/insertions in 17 genes. We identified the TP53 Y102C and NF1 T676fs as putative driver mutations. As the TP53 Y102C was the most abundant mutation identified in all stages, could represent the founding mutation. On the other hand, NF1 T676fs was rapidly enriched in the relapse.Next we delineated a model of clonal evolution using the proportion of SNV and CNA. Although CK with -7 was acquired during disease progression in this case, interestingly, deleted allele of chromosome 7 was different between sAML and relapse phase. In other words, two distinct clones characterized by deletion of different allele (-7A/-7B) existed after sAML, and clonal competition occurred during disease progression and having therapy.(Figure) The sAML stage was characterized by a founding clone harboring TP53 mutation and del(5q), major clone containing -7A and sub-clone with -7B. del(11p) was acquired in -7A clone and became dominant, but in remission the clone with del(11p) was extinct. On the other hand, clonal evolution took place on -7B clone, which acquired NF1 mutation, del(17p) encompassing TP53, and del(17q) including NF1. Finally, the evolved clone from -7B obtained chemo-resistance and contributed to relapse and also -7A clone flared up with deleted NF1.
Conclusion
To analysis serial samples during disease progression discloses that two distinct clone harboring chromosomal aberration on each allele competed with each other by selective pressure of therapy. These observations uncovered the complicated clonal architecture with the accumulations of genetic abnormality.
Session topic: E-poster
Keyword(s): Clonal expansion, MDS/AML
Type: Eposter Presentation
Background
Although myelodysplastic syndromes (MDS) with isolated del(5q) represents a favorable prognosis, the presence of TP53 mutation has been associated with transformation to secondary AML (sAML) accompanied by the emergence of a complex karyotype (CK). The demethylating agent azacitidine (AZA) is currently the standard therapy for patients with high-risk MDS. However, some reports showed AZA treatment cannot improve survival in high-risk MDS and sAML patients with TP53 mutations, in spite of response to AZA. The molecular mechanisms in the acquisition of resistance to AZA in TP53 mutated cases has not been completely understood.
Aims
To clarify the molecular mechanisms underlying disease progression and the acquisition of chemo-resistance in MDS, we delineated clonal evolution during disease course using high throughput sequencing of serial samples.
Methods
The 77-year-old female patient was diagnosed with MDS with isolated del(5q). She then received lenalidomide (LEN), but progressed to sAML with CK involving -7. She received AZA with LEN and then achieved temporarily marrow CR (blast<5%). Although she received same therapies, she relapsed and died of progressive leukemia. Genomic DNA was collected at four stages (RAEB-1, sAML, remission, and relapse) on informed consent. We performed whole exome sequencing (WES) and PCR amplicon based target deep sequencing by NGS (Illumina HiSeq 2500). Copy number alterations (CNA) were analyzed based on the read number and allelic imbalances were estimated from allele frequency of heterozygous SNPs.
Results
The mean coverage of WES and target deep sequencing was 209 times and 67073 times, respectively. We detected 15 somatic single nucleotide variants (SNV) and 2 frameshift/insertions in 17 genes. We identified the TP53 Y102C and NF1 T676fs as putative driver mutations. As the TP53 Y102C was the most abundant mutation identified in all stages, could represent the founding mutation. On the other hand, NF1 T676fs was rapidly enriched in the relapse.Next we delineated a model of clonal evolution using the proportion of SNV and CNA. Although CK with -7 was acquired during disease progression in this case, interestingly, deleted allele of chromosome 7 was different between sAML and relapse phase. In other words, two distinct clones characterized by deletion of different allele (-7A/-7B) existed after sAML, and clonal competition occurred during disease progression and having therapy.(Figure) The sAML stage was characterized by a founding clone harboring TP53 mutation and del(5q), major clone containing -7A and sub-clone with -7B. del(11p) was acquired in -7A clone and became dominant, but in remission the clone with del(11p) was extinct. On the other hand, clonal evolution took place on -7B clone, which acquired NF1 mutation, del(17p) encompassing TP53, and del(17q) including NF1. Finally, the evolved clone from -7B obtained chemo-resistance and contributed to relapse and also -7A clone flared up with deleted NF1.
Conclusion
To analysis serial samples during disease progression discloses that two distinct clone harboring chromosomal aberration on each allele competed with each other by selective pressure of therapy. These observations uncovered the complicated clonal architecture with the accumulations of genetic abnormality.
Session topic: E-poster
Keyword(s): Clonal expansion, MDS/AML
Abstract: E1204
Type: Eposter Presentation
Background
Although myelodysplastic syndromes (MDS) with isolated del(5q) represents a favorable prognosis, the presence of TP53 mutation has been associated with transformation to secondary AML (sAML) accompanied by the emergence of a complex karyotype (CK). The demethylating agent azacitidine (AZA) is currently the standard therapy for patients with high-risk MDS. However, some reports showed AZA treatment cannot improve survival in high-risk MDS and sAML patients with TP53 mutations, in spite of response to AZA. The molecular mechanisms in the acquisition of resistance to AZA in TP53 mutated cases has not been completely understood.
Aims
To clarify the molecular mechanisms underlying disease progression and the acquisition of chemo-resistance in MDS, we delineated clonal evolution during disease course using high throughput sequencing of serial samples.
Methods
The 77-year-old female patient was diagnosed with MDS with isolated del(5q). She then received lenalidomide (LEN), but progressed to sAML with CK involving -7. She received AZA with LEN and then achieved temporarily marrow CR (blast<5%). Although she received same therapies, she relapsed and died of progressive leukemia. Genomic DNA was collected at four stages (RAEB-1, sAML, remission, and relapse) on informed consent. We performed whole exome sequencing (WES) and PCR amplicon based target deep sequencing by NGS (Illumina HiSeq 2500). Copy number alterations (CNA) were analyzed based on the read number and allelic imbalances were estimated from allele frequency of heterozygous SNPs.
Results
The mean coverage of WES and target deep sequencing was 209 times and 67073 times, respectively. We detected 15 somatic single nucleotide variants (SNV) and 2 frameshift/insertions in 17 genes. We identified the TP53 Y102C and NF1 T676fs as putative driver mutations. As the TP53 Y102C was the most abundant mutation identified in all stages, could represent the founding mutation. On the other hand, NF1 T676fs was rapidly enriched in the relapse.Next we delineated a model of clonal evolution using the proportion of SNV and CNA. Although CK with -7 was acquired during disease progression in this case, interestingly, deleted allele of chromosome 7 was different between sAML and relapse phase. In other words, two distinct clones characterized by deletion of different allele (-7A/-7B) existed after sAML, and clonal competition occurred during disease progression and having therapy.(Figure) The sAML stage was characterized by a founding clone harboring TP53 mutation and del(5q), major clone containing -7A and sub-clone with -7B. del(11p) was acquired in -7A clone and became dominant, but in remission the clone with del(11p) was extinct. On the other hand, clonal evolution took place on -7B clone, which acquired NF1 mutation, del(17p) encompassing TP53, and del(17q) including NF1. Finally, the evolved clone from -7B obtained chemo-resistance and contributed to relapse and also -7A clone flared up with deleted NF1.
Conclusion
To analysis serial samples during disease progression discloses that two distinct clone harboring chromosomal aberration on each allele competed with each other by selective pressure of therapy. These observations uncovered the complicated clonal architecture with the accumulations of genetic abnormality.
Session topic: E-poster
Keyword(s): Clonal expansion, MDS/AML
Type: Eposter Presentation
Background
Although myelodysplastic syndromes (MDS) with isolated del(5q) represents a favorable prognosis, the presence of TP53 mutation has been associated with transformation to secondary AML (sAML) accompanied by the emergence of a complex karyotype (CK). The demethylating agent azacitidine (AZA) is currently the standard therapy for patients with high-risk MDS. However, some reports showed AZA treatment cannot improve survival in high-risk MDS and sAML patients with TP53 mutations, in spite of response to AZA. The molecular mechanisms in the acquisition of resistance to AZA in TP53 mutated cases has not been completely understood.
Aims
To clarify the molecular mechanisms underlying disease progression and the acquisition of chemo-resistance in MDS, we delineated clonal evolution during disease course using high throughput sequencing of serial samples.
Methods
The 77-year-old female patient was diagnosed with MDS with isolated del(5q). She then received lenalidomide (LEN), but progressed to sAML with CK involving -7. She received AZA with LEN and then achieved temporarily marrow CR (blast<5%). Although she received same therapies, she relapsed and died of progressive leukemia. Genomic DNA was collected at four stages (RAEB-1, sAML, remission, and relapse) on informed consent. We performed whole exome sequencing (WES) and PCR amplicon based target deep sequencing by NGS (Illumina HiSeq 2500). Copy number alterations (CNA) were analyzed based on the read number and allelic imbalances were estimated from allele frequency of heterozygous SNPs.
Results
The mean coverage of WES and target deep sequencing was 209 times and 67073 times, respectively. We detected 15 somatic single nucleotide variants (SNV) and 2 frameshift/insertions in 17 genes. We identified the TP53 Y102C and NF1 T676fs as putative driver mutations. As the TP53 Y102C was the most abundant mutation identified in all stages, could represent the founding mutation. On the other hand, NF1 T676fs was rapidly enriched in the relapse.Next we delineated a model of clonal evolution using the proportion of SNV and CNA. Although CK with -7 was acquired during disease progression in this case, interestingly, deleted allele of chromosome 7 was different between sAML and relapse phase. In other words, two distinct clones characterized by deletion of different allele (-7A/-7B) existed after sAML, and clonal competition occurred during disease progression and having therapy.(Figure) The sAML stage was characterized by a founding clone harboring TP53 mutation and del(5q), major clone containing -7A and sub-clone with -7B. del(11p) was acquired in -7A clone and became dominant, but in remission the clone with del(11p) was extinct. On the other hand, clonal evolution took place on -7B clone, which acquired NF1 mutation, del(17p) encompassing TP53, and del(17q) including NF1. Finally, the evolved clone from -7B obtained chemo-resistance and contributed to relapse and also -7A clone flared up with deleted NF1.
Conclusion
To analysis serial samples during disease progression discloses that two distinct clone harboring chromosomal aberration on each allele competed with each other by selective pressure of therapy. These observations uncovered the complicated clonal architecture with the accumulations of genetic abnormality.
Session topic: E-poster
Keyword(s): Clonal expansion, MDS/AML
{{ help_message }}
{{filter}}