
EXPANDING THE KNOWLEDGE OF BLOOD TRANSCRIPTOME: CIRCULAR RNAS IN HEMATOPOIESIS
(Abstract release date: 05/19/16)
EHA Library. Bonizzato A. 06/09/16; 132673; E1124

Mrs. Annagiulia Bonizzato
Contributions
Contributions
Abstract
Abstract: E1124
Type: Eposter Presentation
Background
Circular RNAs (circRNAs) are a class of covalently closed RNA molecules generated by back-splicing joining exons in a non-collinear way. circRNAs have been found throughout the tree of life and are conserved in mammals. As reported circRNAs may decoy miRNAs thus de-repressing miRNA targets and regulating cellular processes and may interact with RNA-binding proteins and other RNAs. High stability and complex cell type- and differentiation-specific expression patterns make circRNAs promising disease biomarkers. In cancer circRNAs are highly relevant since they associate with cancer miRNAs and regulate cancer-related pathways. Moreover, specific circRNAs exhibit anti-cancer effects, whereas others discriminate malignant cells from healthy ones. Recent studies demonstrated that the circRNA number and expression level in normal blood is reproducible and higher than in other tissues and incipient observations detected circRNAs in B-ALL.
Aims
We aim to detect, discover and characterize circRNAs expressed during hematopoiesis in order to expand the knowledge of transcriptome complexity and variations in normal differentiation to set the basis for circRNA studies in hematologic malignancies.
Methods
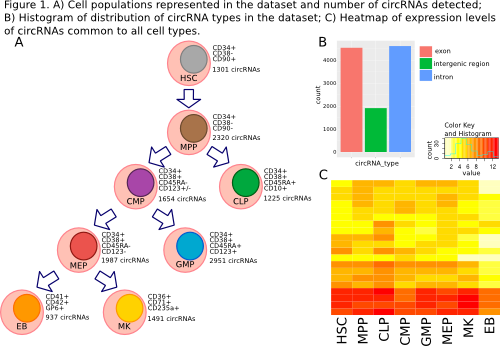
CircRNA detection grounds on identification of RNA-seq reads that contain a back-splice junction. Different programs rely on slightly different approaches, such as using a pre-compiled dataset of all possible exons of each gene coupled in reverse order, or by de novo identifying the back-splice junction from signals in the alignment data coupled with signals of canonical splicing. Combining the output of at least two programs increases the accuracy of true circRNA detection, as suggested by recent literature. Poly-A enriched RNA-seq data of eight cell populations of the hematopoietic tree (Figure 1A) were collected (HSC, MPP, CMP, CLP, GMP, MEP, MK, EB cells; three biological replicates per cell type; ID: EGAS00001000284, by Chen L et al., 2014, Science).
Results
A bioinformatics pipeline for detection and analysis of cirRNAs from RNA-seq data was set up that brings together published circRNA detection tools (CIRI, find_circ and testrealign.x) and custom made scripts, with a modular design expandable to novel programs. Preliminary analyses performed with CIRI allowed us to detect 11,079 putative circRNAs expressed during hematopoiesis, of which 41% are exonic, 42% are intronic and 17% fall in regions annotated as intergenic (Figure 1B). Expressed circRNAs are associated to 4,317 known genes, 58% of which produce only one circRNA, whereas 837 (19%) genes produce two different circular isoforms each, and 959 (22%) even more than three isoforms.The large majority (87.5%) of circRNAs was detected only in one cell type, and in each cell population on average 15% of the total detected circRNAs is present. We identified 20 circRNAs expressed in all cell stages (Figure 1C) and defined several patterns of circRNAs that are up or down regulated in less differentiated cells compared to more mature stages (e.g. HSC vs CMP), specifically in the lymphoid compared with the myeloid branch of the tree (CMP vs CLP) and, through the myeloid lineage, from CMP to more and terminally differentiated cells.
Conclusion
Recent studies have unveiled a great potential of circRNAs in hematology. Our results, although preliminary, confirm that a high number of circRNAs are expressed during hematopoietic cell differentiation. We identified circRNAs expressed by all cell stages in the dataset, and several expression patterns that seem to be specific of stem cells, of differentiated cells, or of cellular lineages. We plan to conduct additional RNA-seq data analyses with different approaches to enrich the set of detected circRNAs in true positive hits, in order to validate them, to confirm some of the more striking expression variations observed and to predict functions and interactions of expressed circRNAs.
Session topic: E-poster
Keyword(s): Alternative splicing, Gene expression, Hematopoiesis, Molecular markers
Type: Eposter Presentation
Background
Circular RNAs (circRNAs) are a class of covalently closed RNA molecules generated by back-splicing joining exons in a non-collinear way. circRNAs have been found throughout the tree of life and are conserved in mammals. As reported circRNAs may decoy miRNAs thus de-repressing miRNA targets and regulating cellular processes and may interact with RNA-binding proteins and other RNAs. High stability and complex cell type- and differentiation-specific expression patterns make circRNAs promising disease biomarkers. In cancer circRNAs are highly relevant since they associate with cancer miRNAs and regulate cancer-related pathways. Moreover, specific circRNAs exhibit anti-cancer effects, whereas others discriminate malignant cells from healthy ones. Recent studies demonstrated that the circRNA number and expression level in normal blood is reproducible and higher than in other tissues and incipient observations detected circRNAs in B-ALL.
Aims
We aim to detect, discover and characterize circRNAs expressed during hematopoiesis in order to expand the knowledge of transcriptome complexity and variations in normal differentiation to set the basis for circRNA studies in hematologic malignancies.
Methods
CircRNA detection grounds on identification of RNA-seq reads that contain a back-splice junction. Different programs rely on slightly different approaches, such as using a pre-compiled dataset of all possible exons of each gene coupled in reverse order, or by de novo identifying the back-splice junction from signals in the alignment data coupled with signals of canonical splicing. Combining the output of at least two programs increases the accuracy of true circRNA detection, as suggested by recent literature. Poly-A enriched RNA-seq data of eight cell populations of the hematopoietic tree (Figure 1A) were collected (HSC, MPP, CMP, CLP, GMP, MEP, MK, EB cells; three biological replicates per cell type; ID: EGAS00001000284, by Chen L et al., 2014, Science).
Results
A bioinformatics pipeline for detection and analysis of cirRNAs from RNA-seq data was set up that brings together published circRNA detection tools (CIRI, find_circ and testrealign.x) and custom made scripts, with a modular design expandable to novel programs. Preliminary analyses performed with CIRI allowed us to detect 11,079 putative circRNAs expressed during hematopoiesis, of which 41% are exonic, 42% are intronic and 17% fall in regions annotated as intergenic (Figure 1B). Expressed circRNAs are associated to 4,317 known genes, 58% of which produce only one circRNA, whereas 837 (19%) genes produce two different circular isoforms each, and 959 (22%) even more than three isoforms.The large majority (87.5%) of circRNAs was detected only in one cell type, and in each cell population on average 15% of the total detected circRNAs is present. We identified 20 circRNAs expressed in all cell stages (Figure 1C) and defined several patterns of circRNAs that are up or down regulated in less differentiated cells compared to more mature stages (e.g. HSC vs CMP), specifically in the lymphoid compared with the myeloid branch of the tree (CMP vs CLP) and, through the myeloid lineage, from CMP to more and terminally differentiated cells.
Conclusion
Recent studies have unveiled a great potential of circRNAs in hematology. Our results, although preliminary, confirm that a high number of circRNAs are expressed during hematopoietic cell differentiation. We identified circRNAs expressed by all cell stages in the dataset, and several expression patterns that seem to be specific of stem cells, of differentiated cells, or of cellular lineages. We plan to conduct additional RNA-seq data analyses with different approaches to enrich the set of detected circRNAs in true positive hits, in order to validate them, to confirm some of the more striking expression variations observed and to predict functions and interactions of expressed circRNAs.
Session topic: E-poster
Keyword(s): Alternative splicing, Gene expression, Hematopoiesis, Molecular markers
Abstract: E1124
Type: Eposter Presentation
Background
Circular RNAs (circRNAs) are a class of covalently closed RNA molecules generated by back-splicing joining exons in a non-collinear way. circRNAs have been found throughout the tree of life and are conserved in mammals. As reported circRNAs may decoy miRNAs thus de-repressing miRNA targets and regulating cellular processes and may interact with RNA-binding proteins and other RNAs. High stability and complex cell type- and differentiation-specific expression patterns make circRNAs promising disease biomarkers. In cancer circRNAs are highly relevant since they associate with cancer miRNAs and regulate cancer-related pathways. Moreover, specific circRNAs exhibit anti-cancer effects, whereas others discriminate malignant cells from healthy ones. Recent studies demonstrated that the circRNA number and expression level in normal blood is reproducible and higher than in other tissues and incipient observations detected circRNAs in B-ALL.
Aims
We aim to detect, discover and characterize circRNAs expressed during hematopoiesis in order to expand the knowledge of transcriptome complexity and variations in normal differentiation to set the basis for circRNA studies in hematologic malignancies.
Methods
CircRNA detection grounds on identification of RNA-seq reads that contain a back-splice junction. Different programs rely on slightly different approaches, such as using a pre-compiled dataset of all possible exons of each gene coupled in reverse order, or by de novo identifying the back-splice junction from signals in the alignment data coupled with signals of canonical splicing. Combining the output of at least two programs increases the accuracy of true circRNA detection, as suggested by recent literature. Poly-A enriched RNA-seq data of eight cell populations of the hematopoietic tree (Figure 1A) were collected (HSC, MPP, CMP, CLP, GMP, MEP, MK, EB cells; three biological replicates per cell type; ID: EGAS00001000284, by Chen L et al., 2014, Science).
Results
A bioinformatics pipeline for detection and analysis of cirRNAs from RNA-seq data was set up that brings together published circRNA detection tools (CIRI, find_circ and testrealign.x) and custom made scripts, with a modular design expandable to novel programs. Preliminary analyses performed with CIRI allowed us to detect 11,079 putative circRNAs expressed during hematopoiesis, of which 41% are exonic, 42% are intronic and 17% fall in regions annotated as intergenic (Figure 1B). Expressed circRNAs are associated to 4,317 known genes, 58% of which produce only one circRNA, whereas 837 (19%) genes produce two different circular isoforms each, and 959 (22%) even more than three isoforms.The large majority (87.5%) of circRNAs was detected only in one cell type, and in each cell population on average 15% of the total detected circRNAs is present. We identified 20 circRNAs expressed in all cell stages (Figure 1C) and defined several patterns of circRNAs that are up or down regulated in less differentiated cells compared to more mature stages (e.g. HSC vs CMP), specifically in the lymphoid compared with the myeloid branch of the tree (CMP vs CLP) and, through the myeloid lineage, from CMP to more and terminally differentiated cells.
Conclusion
Recent studies have unveiled a great potential of circRNAs in hematology. Our results, although preliminary, confirm that a high number of circRNAs are expressed during hematopoietic cell differentiation. We identified circRNAs expressed by all cell stages in the dataset, and several expression patterns that seem to be specific of stem cells, of differentiated cells, or of cellular lineages. We plan to conduct additional RNA-seq data analyses with different approaches to enrich the set of detected circRNAs in true positive hits, in order to validate them, to confirm some of the more striking expression variations observed and to predict functions and interactions of expressed circRNAs.
Session topic: E-poster
Keyword(s): Alternative splicing, Gene expression, Hematopoiesis, Molecular markers
Type: Eposter Presentation
Background
Circular RNAs (circRNAs) are a class of covalently closed RNA molecules generated by back-splicing joining exons in a non-collinear way. circRNAs have been found throughout the tree of life and are conserved in mammals. As reported circRNAs may decoy miRNAs thus de-repressing miRNA targets and regulating cellular processes and may interact with RNA-binding proteins and other RNAs. High stability and complex cell type- and differentiation-specific expression patterns make circRNAs promising disease biomarkers. In cancer circRNAs are highly relevant since they associate with cancer miRNAs and regulate cancer-related pathways. Moreover, specific circRNAs exhibit anti-cancer effects, whereas others discriminate malignant cells from healthy ones. Recent studies demonstrated that the circRNA number and expression level in normal blood is reproducible and higher than in other tissues and incipient observations detected circRNAs in B-ALL.
Aims
We aim to detect, discover and characterize circRNAs expressed during hematopoiesis in order to expand the knowledge of transcriptome complexity and variations in normal differentiation to set the basis for circRNA studies in hematologic malignancies.
Methods
CircRNA detection grounds on identification of RNA-seq reads that contain a back-splice junction. Different programs rely on slightly different approaches, such as using a pre-compiled dataset of all possible exons of each gene coupled in reverse order, or by de novo identifying the back-splice junction from signals in the alignment data coupled with signals of canonical splicing. Combining the output of at least two programs increases the accuracy of true circRNA detection, as suggested by recent literature. Poly-A enriched RNA-seq data of eight cell populations of the hematopoietic tree (Figure 1A) were collected (HSC, MPP, CMP, CLP, GMP, MEP, MK, EB cells; three biological replicates per cell type; ID: EGAS00001000284, by Chen L et al., 2014, Science).
Results
A bioinformatics pipeline for detection and analysis of cirRNAs from RNA-seq data was set up that brings together published circRNA detection tools (CIRI, find_circ and testrealign.x) and custom made scripts, with a modular design expandable to novel programs. Preliminary analyses performed with CIRI allowed us to detect 11,079 putative circRNAs expressed during hematopoiesis, of which 41% are exonic, 42% are intronic and 17% fall in regions annotated as intergenic (Figure 1B). Expressed circRNAs are associated to 4,317 known genes, 58% of which produce only one circRNA, whereas 837 (19%) genes produce two different circular isoforms each, and 959 (22%) even more than three isoforms.The large majority (87.5%) of circRNAs was detected only in one cell type, and in each cell population on average 15% of the total detected circRNAs is present. We identified 20 circRNAs expressed in all cell stages (Figure 1C) and defined several patterns of circRNAs that are up or down regulated in less differentiated cells compared to more mature stages (e.g. HSC vs CMP), specifically in the lymphoid compared with the myeloid branch of the tree (CMP vs CLP) and, through the myeloid lineage, from CMP to more and terminally differentiated cells.
Conclusion
Recent studies have unveiled a great potential of circRNAs in hematology. Our results, although preliminary, confirm that a high number of circRNAs are expressed during hematopoietic cell differentiation. We identified circRNAs expressed by all cell stages in the dataset, and several expression patterns that seem to be specific of stem cells, of differentiated cells, or of cellular lineages. We plan to conduct additional RNA-seq data analyses with different approaches to enrich the set of detected circRNAs in true positive hits, in order to validate them, to confirm some of the more striking expression variations observed and to predict functions and interactions of expressed circRNAs.
Session topic: E-poster
Keyword(s): Alternative splicing, Gene expression, Hematopoiesis, Molecular markers
{{ help_message }}
{{filter}}