
PREVALENCE OF AGE-RELATED CLONAL HEMATOPOIESIS (ARCH) AND LOW FREQUENCY MUTATIONS IN CLL & MONOCLONAL B LYMPHOCYTOSIS: A MAYO CLINIC EXOME SEQUENCING COHORT STUDY
(Abstract release date: 05/19/16)
EHA Library. Foran J. 06/09/16; 132620; E1071
Disclosure(s): NO RELEVANT FINANCIAL DISCLOSURES
FUNDED BY MAYO CLINIC FLORIDA CRT-2 AWARD [J. FORAN, PI]

Dr. James M Foran
Contributions
Contributions
Abstract
Abstract: E1071
Type: Eposter Presentation
Background
ARCH refers to somatically acquired single-nucleotide variants (SNV's) and small indels in DNA involving hematologic (and non-hematologic) genes (most commonly DNMT3A, TET2, ASXL1 & PPM1D) detected by whole-exome sequencing (WES) in peripheral blood in unaffected individuals. The incidence of ARCH increases with age and is asociated with an 11-fold increase in the risk of hematologic malignancy including leukemia. However, the contribution of ARCH to the development of CLL, the hematologic malignancy with highest prevalence, is unknown. Furthermore, the frequency and clinical impact of ARCH-associated and other non-ARCH low frequency mutations is unknown in CLL. We therefore sought to understand the contibution and frequency of ARCH and other low frequency mutations in a cohort of patients with CLL and monoclonal B-lymphocytosis (MBL, an established precursor state with increased risk of CLL development) who have undergone WES.
Aims
To determine the prevalence of ARCH and non-ARCH low frequency mutations and their association with clinical risk factors in a cohort of unaffected (UA) and affected individuals with CLL & MBL using peripheral blood WES.
Methods
Bioinformatic analysis was performed using raw data (BAM and FASTQ files) from WES performed in a cohort of 445 patients, comprised of CLL (n=160), MBL (n=73), and unaffected individuals (UA, n=212). Samples were prepared using the Agilent V2 Exome Capture Kit, and in some cases prepared using the Agilent V4 Exome Capture Kit. WES alignment was performed with Novoalign within GGPS and recalibrated and realigned with GATK. Pileup files were obtained from the recalibrated and realigned BAM files, and identification of clonal and sub-clonal variants performed using DV-Boosting method and Q value of 0.05 as cut off. Consistent with previous evaluation of ARCH, “non-functional” variants were filtered out (synonymous, UTR, intron, etc.). In addition, variants with an Alternative Allele Frequeny (AAF) ≥0.05 in European or Caucasian populations of HapMap, 1000G, and ESP6500 were filtered out as 'polymorphisms' and not included as mutations. Remaining variants were categorized as “disruptive” and “non-synonymous” (NS) variants in accordance with previous publications. Disruptive SNV's included those with Start/Stop codon and at splice sites. Because of the inclusion of 'affected' individuals in this analysis who may have subclinical clonal disease variants, we defined the “non-inherited” variants more conservatively as those with <40% of the AAF.
Results
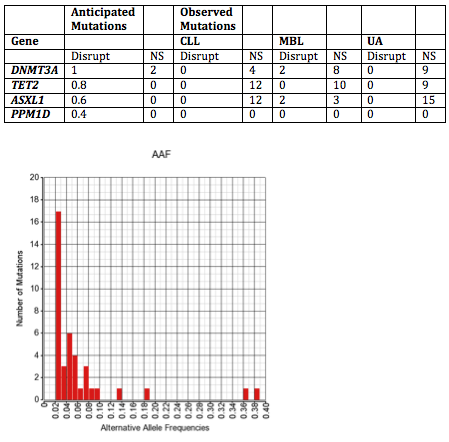
The frequency of detectable mutations is noted in Figure. In almost all cases the AAF was ≤10%. Based on previous population-based studies, we anticipated a rate of disruptive (Disrupt) and NS ARCH mutations for the cohort of 445 patients (noted in Table). We did not detect ARCH mutations in any CLL or UA patient, and in total detected DNMT3A and ASXL1 mutations each in 2 MBL patients. We detected non-ARCH low AAF disruptive somatic mutations in other genes in 12 CLL and 11 MBL patients, respectively, including in 4 Tier 1 cancer genes (JAK2, FGFR3, MLH1 & STK11) occurring in 3 patients each with CLL and MBL. No mutations were detected in UA population in this analysis.To determine if low AAF mutations were associated with any unique CLL clinical characteristics, we compared frequencies among the subset of CLL patients. Among those with available Rai stage, CLL patients with a detectable low AAF mutation appeared to be more likely to have advanced (stage III/IV) disease at diagnosis (25%) compared with those without a mutations (2.3%) (p=0.03), but there was no difference in early stage disease (Stage 0: 75% vs. 67.7%; and Stage I/II: 0% vs. 31.1%, respectively) (p=0.13). There was no difference in age at diagnosis or gender.
Conclusion
ARCH appears to be a very infrequent event in CLL, and therefore is unlikely to constitute a major risk factor for CLL development. Low AAF non-ARCH mutations are detectable in some patients with CLL (7.5% in this analysis), including in some Tier 1 cancer genes, and may be associated with advanced stage disease at diagnosis, although this is based on a limited number of observations and will require prospective confirmation. These results suggest an important role for Deep Sequencing in advanced CLL focusing on Tier 1 cancer genes.
Session topic: E-poster
Keyword(s): Chronic lymphocytic leukemia, Clonality, Hematopoiesis
Type: Eposter Presentation
Background
ARCH refers to somatically acquired single-nucleotide variants (SNV's) and small indels in DNA involving hematologic (and non-hematologic) genes (most commonly DNMT3A, TET2, ASXL1 & PPM1D) detected by whole-exome sequencing (WES) in peripheral blood in unaffected individuals. The incidence of ARCH increases with age and is asociated with an 11-fold increase in the risk of hematologic malignancy including leukemia. However, the contribution of ARCH to the development of CLL, the hematologic malignancy with highest prevalence, is unknown. Furthermore, the frequency and clinical impact of ARCH-associated and other non-ARCH low frequency mutations is unknown in CLL. We therefore sought to understand the contibution and frequency of ARCH and other low frequency mutations in a cohort of patients with CLL and monoclonal B-lymphocytosis (MBL, an established precursor state with increased risk of CLL development) who have undergone WES.
Aims
To determine the prevalence of ARCH and non-ARCH low frequency mutations and their association with clinical risk factors in a cohort of unaffected (UA) and affected individuals with CLL & MBL using peripheral blood WES.
Methods
Bioinformatic analysis was performed using raw data (BAM and FASTQ files) from WES performed in a cohort of 445 patients, comprised of CLL (n=160), MBL (n=73), and unaffected individuals (UA, n=212). Samples were prepared using the Agilent V2 Exome Capture Kit, and in some cases prepared using the Agilent V4 Exome Capture Kit. WES alignment was performed with Novoalign within GGPS and recalibrated and realigned with GATK. Pileup files were obtained from the recalibrated and realigned BAM files, and identification of clonal and sub-clonal variants performed using DV-Boosting method and Q value of 0.05 as cut off. Consistent with previous evaluation of ARCH, “non-functional” variants were filtered out (synonymous, UTR, intron, etc.). In addition, variants with an Alternative Allele Frequeny (AAF) ≥0.05 in European or Caucasian populations of HapMap, 1000G, and ESP6500 were filtered out as 'polymorphisms' and not included as mutations. Remaining variants were categorized as “disruptive” and “non-synonymous” (NS) variants in accordance with previous publications. Disruptive SNV's included those with Start/Stop codon and at splice sites. Because of the inclusion of 'affected' individuals in this analysis who may have subclinical clonal disease variants, we defined the “non-inherited” variants more conservatively as those with <40% of the AAF.
Results
The frequency of detectable mutations is noted in Figure. In almost all cases the AAF was ≤10%. Based on previous population-based studies, we anticipated a rate of disruptive (Disrupt) and NS ARCH mutations for the cohort of 445 patients (noted in Table). We did not detect ARCH mutations in any CLL or UA patient, and in total detected DNMT3A and ASXL1 mutations each in 2 MBL patients. We detected non-ARCH low AAF disruptive somatic mutations in other genes in 12 CLL and 11 MBL patients, respectively, including in 4 Tier 1 cancer genes (JAK2, FGFR3, MLH1 & STK11) occurring in 3 patients each with CLL and MBL. No mutations were detected in UA population in this analysis.To determine if low AAF mutations were associated with any unique CLL clinical characteristics, we compared frequencies among the subset of CLL patients. Among those with available Rai stage, CLL patients with a detectable low AAF mutation appeared to be more likely to have advanced (stage III/IV) disease at diagnosis (25%) compared with those without a mutations (2.3%) (p=0.03), but there was no difference in early stage disease (Stage 0: 75% vs. 67.7%; and Stage I/II: 0% vs. 31.1%, respectively) (p=0.13). There was no difference in age at diagnosis or gender.
Conclusion
ARCH appears to be a very infrequent event in CLL, and therefore is unlikely to constitute a major risk factor for CLL development. Low AAF non-ARCH mutations are detectable in some patients with CLL (7.5% in this analysis), including in some Tier 1 cancer genes, and may be associated with advanced stage disease at diagnosis, although this is based on a limited number of observations and will require prospective confirmation. These results suggest an important role for Deep Sequencing in advanced CLL focusing on Tier 1 cancer genes.
Session topic: E-poster
Keyword(s): Chronic lymphocytic leukemia, Clonality, Hematopoiesis
Abstract: E1071
Type: Eposter Presentation
Background
ARCH refers to somatically acquired single-nucleotide variants (SNV's) and small indels in DNA involving hematologic (and non-hematologic) genes (most commonly DNMT3A, TET2, ASXL1 & PPM1D) detected by whole-exome sequencing (WES) in peripheral blood in unaffected individuals. The incidence of ARCH increases with age and is asociated with an 11-fold increase in the risk of hematologic malignancy including leukemia. However, the contribution of ARCH to the development of CLL, the hematologic malignancy with highest prevalence, is unknown. Furthermore, the frequency and clinical impact of ARCH-associated and other non-ARCH low frequency mutations is unknown in CLL. We therefore sought to understand the contibution and frequency of ARCH and other low frequency mutations in a cohort of patients with CLL and monoclonal B-lymphocytosis (MBL, an established precursor state with increased risk of CLL development) who have undergone WES.
Aims
To determine the prevalence of ARCH and non-ARCH low frequency mutations and their association with clinical risk factors in a cohort of unaffected (UA) and affected individuals with CLL & MBL using peripheral blood WES.
Methods
Bioinformatic analysis was performed using raw data (BAM and FASTQ files) from WES performed in a cohort of 445 patients, comprised of CLL (n=160), MBL (n=73), and unaffected individuals (UA, n=212). Samples were prepared using the Agilent V2 Exome Capture Kit, and in some cases prepared using the Agilent V4 Exome Capture Kit. WES alignment was performed with Novoalign within GGPS and recalibrated and realigned with GATK. Pileup files were obtained from the recalibrated and realigned BAM files, and identification of clonal and sub-clonal variants performed using DV-Boosting method and Q value of 0.05 as cut off. Consistent with previous evaluation of ARCH, “non-functional” variants were filtered out (synonymous, UTR, intron, etc.). In addition, variants with an Alternative Allele Frequeny (AAF) ≥0.05 in European or Caucasian populations of HapMap, 1000G, and ESP6500 were filtered out as 'polymorphisms' and not included as mutations. Remaining variants were categorized as “disruptive” and “non-synonymous” (NS) variants in accordance with previous publications. Disruptive SNV's included those with Start/Stop codon and at splice sites. Because of the inclusion of 'affected' individuals in this analysis who may have subclinical clonal disease variants, we defined the “non-inherited” variants more conservatively as those with <40% of the AAF.
Results
The frequency of detectable mutations is noted in Figure. In almost all cases the AAF was ≤10%. Based on previous population-based studies, we anticipated a rate of disruptive (Disrupt) and NS ARCH mutations for the cohort of 445 patients (noted in Table). We did not detect ARCH mutations in any CLL or UA patient, and in total detected DNMT3A and ASXL1 mutations each in 2 MBL patients. We detected non-ARCH low AAF disruptive somatic mutations in other genes in 12 CLL and 11 MBL patients, respectively, including in 4 Tier 1 cancer genes (JAK2, FGFR3, MLH1 & STK11) occurring in 3 patients each with CLL and MBL. No mutations were detected in UA population in this analysis.To determine if low AAF mutations were associated with any unique CLL clinical characteristics, we compared frequencies among the subset of CLL patients. Among those with available Rai stage, CLL patients with a detectable low AAF mutation appeared to be more likely to have advanced (stage III/IV) disease at diagnosis (25%) compared with those without a mutations (2.3%) (p=0.03), but there was no difference in early stage disease (Stage 0: 75% vs. 67.7%; and Stage I/II: 0% vs. 31.1%, respectively) (p=0.13). There was no difference in age at diagnosis or gender.
Conclusion
ARCH appears to be a very infrequent event in CLL, and therefore is unlikely to constitute a major risk factor for CLL development. Low AAF non-ARCH mutations are detectable in some patients with CLL (7.5% in this analysis), including in some Tier 1 cancer genes, and may be associated with advanced stage disease at diagnosis, although this is based on a limited number of observations and will require prospective confirmation. These results suggest an important role for Deep Sequencing in advanced CLL focusing on Tier 1 cancer genes.
Session topic: E-poster
Keyword(s): Chronic lymphocytic leukemia, Clonality, Hematopoiesis
Type: Eposter Presentation
Background
ARCH refers to somatically acquired single-nucleotide variants (SNV's) and small indels in DNA involving hematologic (and non-hematologic) genes (most commonly DNMT3A, TET2, ASXL1 & PPM1D) detected by whole-exome sequencing (WES) in peripheral blood in unaffected individuals. The incidence of ARCH increases with age and is asociated with an 11-fold increase in the risk of hematologic malignancy including leukemia. However, the contribution of ARCH to the development of CLL, the hematologic malignancy with highest prevalence, is unknown. Furthermore, the frequency and clinical impact of ARCH-associated and other non-ARCH low frequency mutations is unknown in CLL. We therefore sought to understand the contibution and frequency of ARCH and other low frequency mutations in a cohort of patients with CLL and monoclonal B-lymphocytosis (MBL, an established precursor state with increased risk of CLL development) who have undergone WES.
Aims
To determine the prevalence of ARCH and non-ARCH low frequency mutations and their association with clinical risk factors in a cohort of unaffected (UA) and affected individuals with CLL & MBL using peripheral blood WES.
Methods
Bioinformatic analysis was performed using raw data (BAM and FASTQ files) from WES performed in a cohort of 445 patients, comprised of CLL (n=160), MBL (n=73), and unaffected individuals (UA, n=212). Samples were prepared using the Agilent V2 Exome Capture Kit, and in some cases prepared using the Agilent V4 Exome Capture Kit. WES alignment was performed with Novoalign within GGPS and recalibrated and realigned with GATK. Pileup files were obtained from the recalibrated and realigned BAM files, and identification of clonal and sub-clonal variants performed using DV-Boosting method and Q value of 0.05 as cut off. Consistent with previous evaluation of ARCH, “non-functional” variants were filtered out (synonymous, UTR, intron, etc.). In addition, variants with an Alternative Allele Frequeny (AAF) ≥0.05 in European or Caucasian populations of HapMap, 1000G, and ESP6500 were filtered out as 'polymorphisms' and not included as mutations. Remaining variants were categorized as “disruptive” and “non-synonymous” (NS) variants in accordance with previous publications. Disruptive SNV's included those with Start/Stop codon and at splice sites. Because of the inclusion of 'affected' individuals in this analysis who may have subclinical clonal disease variants, we defined the “non-inherited” variants more conservatively as those with <40% of the AAF.
Results
The frequency of detectable mutations is noted in Figure. In almost all cases the AAF was ≤10%. Based on previous population-based studies, we anticipated a rate of disruptive (Disrupt) and NS ARCH mutations for the cohort of 445 patients (noted in Table). We did not detect ARCH mutations in any CLL or UA patient, and in total detected DNMT3A and ASXL1 mutations each in 2 MBL patients. We detected non-ARCH low AAF disruptive somatic mutations in other genes in 12 CLL and 11 MBL patients, respectively, including in 4 Tier 1 cancer genes (JAK2, FGFR3, MLH1 & STK11) occurring in 3 patients each with CLL and MBL. No mutations were detected in UA population in this analysis.To determine if low AAF mutations were associated with any unique CLL clinical characteristics, we compared frequencies among the subset of CLL patients. Among those with available Rai stage, CLL patients with a detectable low AAF mutation appeared to be more likely to have advanced (stage III/IV) disease at diagnosis (25%) compared with those without a mutations (2.3%) (p=0.03), but there was no difference in early stage disease (Stage 0: 75% vs. 67.7%; and Stage I/II: 0% vs. 31.1%, respectively) (p=0.13). There was no difference in age at diagnosis or gender.
Conclusion
ARCH appears to be a very infrequent event in CLL, and therefore is unlikely to constitute a major risk factor for CLL development. Low AAF non-ARCH mutations are detectable in some patients with CLL (7.5% in this analysis), including in some Tier 1 cancer genes, and may be associated with advanced stage disease at diagnosis, although this is based on a limited number of observations and will require prospective confirmation. These results suggest an important role for Deep Sequencing in advanced CLL focusing on Tier 1 cancer genes.
Session topic: E-poster
Keyword(s): Chronic lymphocytic leukemia, Clonality, Hematopoiesis
{{ help_message }}
{{filter}}