
LINKING PROTEOME AND GENOME FOR BIOMARKER DISCOVERY IN CLL – ESTABLISHMENT OF A CLL PROTEIN DATABASE
(Abstract release date: 05/19/16)
EHA Library. Eagle G. 06/09/16; 132571; E1022

Dr. Gina Eagle
Contributions
Contributions
Abstract
Abstract: E1022
Type: Eposter Presentation
Background
Chronic Lymphocytic Leukaemia (CLL) is notable for its clinical variability, suggesting a role for precision medicine. Attempts at understanding the biological basis of this variability have mainly focussed on genomic alterations and gene expression at the mRNA level. However, the molecular basis of CLL variability remains incompletely understood. We speculate that this is because the clinical phenotype is ultimately determined by gene expression at the protein level. We have therefore embarked on a global proteomic study of a large number of CLL trial samples for which whole genome sequencing data will be available through the Genomics England Ltd (GEL) CLL Pilot Project. We anticipate that our study will bridge the gap in our understanding between genotype and clinical phenotype.
Aims
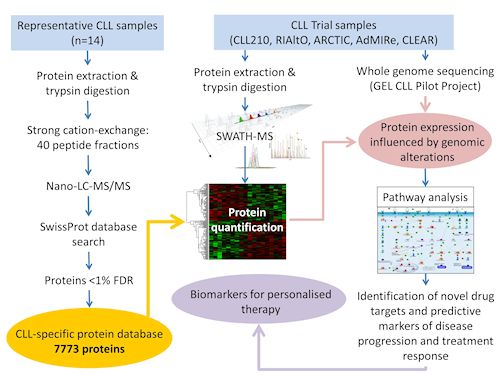
Recently developed mass spectrometric approaches to highly parallel, label-free protein quantification provide a new opportunity to develop diagnostic, prognostic and predictive biomarkers. SWATH (Sequential Windowed Acquisition of all THeoretical fragments) is a data-independent approach which generates a mass spectral library of fragment ions from all detectable peptide precursors. The composite MS/MS spectra are deconvoluted by alignment with a high quality and exhaustive, tissue specific database, whereupon patient samples may be stratified based on the quantitative expression profile of thousands of proteins. Here, we describe generation of a CLL-specific protein database that will provide a permanent reference source for all future trial samples, as indicated in Figure 1.
Methods
Protein extracts were prepared from CLL cells from 14 individual patients in various stages of the disease. Lysates were pooled and digested before being subjected to cation exchange chromatography. Forty fractions were delivered into a TripleTOF 6600 mass spectrometer (SCIEX) via an Eksigent nanoLC 415 system (SCIEX). Data dependent acquisition was performed using 25 MS/MS per cycle to optimise for quality and 30 MS/MS per cycle to optimise for coverage, and the combined data were searched using ProteinPilot 5.0 (SCIEX) against the SwissProt database (Nov 2015, 20,193 human entries). Proteins lying within a 1% global false discovery rate were included in the database.
Results
The new CLL-specific database contains 7773 proteins. The database covers 50% of all human UniProtKB/SwissProt entries which have evidence at the protein level. Pathway analysis of B cell receptor (BCR) signalling based on the GeneGo pathway maps in the MetaCore database (version 6.14, build 61508; Thomson Reuters) showed that this CLL database covers over 87% of the proteins involved in BCR signalling. The quality of the database entries is further underlined by high representation in the Human Genome database (20,814 entries) in aspect of Gene Ontology biological processes and molecular functions, as determined by PANTHER over-representation tests.
Conclusion
Our global protein expression database for CLL cells is thus the most complete database of its type. By combining advanced computational biology to relate protein expression to whole genome sequencing data, the database will provide a framework for elucidating the effect of genomic alterations on protein expression. The construction of this comprehensive CLL database also provides the platform for applying SWATH/MS-based analysis of individual patient samples to identify novel drug targets and predictive biomarkers of disease progression and treatment response.
Session topic: E-poster
Keyword(s): Chronic lymphocytic leukemia, Proteomics
Type: Eposter Presentation
Background
Chronic Lymphocytic Leukaemia (CLL) is notable for its clinical variability, suggesting a role for precision medicine. Attempts at understanding the biological basis of this variability have mainly focussed on genomic alterations and gene expression at the mRNA level. However, the molecular basis of CLL variability remains incompletely understood. We speculate that this is because the clinical phenotype is ultimately determined by gene expression at the protein level. We have therefore embarked on a global proteomic study of a large number of CLL trial samples for which whole genome sequencing data will be available through the Genomics England Ltd (GEL) CLL Pilot Project. We anticipate that our study will bridge the gap in our understanding between genotype and clinical phenotype.
Aims
Recently developed mass spectrometric approaches to highly parallel, label-free protein quantification provide a new opportunity to develop diagnostic, prognostic and predictive biomarkers. SWATH (Sequential Windowed Acquisition of all THeoretical fragments) is a data-independent approach which generates a mass spectral library of fragment ions from all detectable peptide precursors. The composite MS/MS spectra are deconvoluted by alignment with a high quality and exhaustive, tissue specific database, whereupon patient samples may be stratified based on the quantitative expression profile of thousands of proteins. Here, we describe generation of a CLL-specific protein database that will provide a permanent reference source for all future trial samples, as indicated in Figure 1.
Methods
Protein extracts were prepared from CLL cells from 14 individual patients in various stages of the disease. Lysates were pooled and digested before being subjected to cation exchange chromatography. Forty fractions were delivered into a TripleTOF 6600 mass spectrometer (SCIEX) via an Eksigent nanoLC 415 system (SCIEX). Data dependent acquisition was performed using 25 MS/MS per cycle to optimise for quality and 30 MS/MS per cycle to optimise for coverage, and the combined data were searched using ProteinPilot 5.0 (SCIEX) against the SwissProt database (Nov 2015, 20,193 human entries). Proteins lying within a 1% global false discovery rate were included in the database.
Results
The new CLL-specific database contains 7773 proteins. The database covers 50% of all human UniProtKB/SwissProt entries which have evidence at the protein level. Pathway analysis of B cell receptor (BCR) signalling based on the GeneGo pathway maps in the MetaCore database (version 6.14, build 61508; Thomson Reuters) showed that this CLL database covers over 87% of the proteins involved in BCR signalling. The quality of the database entries is further underlined by high representation in the Human Genome database (20,814 entries) in aspect of Gene Ontology biological processes and molecular functions, as determined by PANTHER over-representation tests.
Conclusion
Our global protein expression database for CLL cells is thus the most complete database of its type. By combining advanced computational biology to relate protein expression to whole genome sequencing data, the database will provide a framework for elucidating the effect of genomic alterations on protein expression. The construction of this comprehensive CLL database also provides the platform for applying SWATH/MS-based analysis of individual patient samples to identify novel drug targets and predictive biomarkers of disease progression and treatment response.
Session topic: E-poster
Keyword(s): Chronic lymphocytic leukemia, Proteomics
Abstract: E1022
Type: Eposter Presentation
Background
Chronic Lymphocytic Leukaemia (CLL) is notable for its clinical variability, suggesting a role for precision medicine. Attempts at understanding the biological basis of this variability have mainly focussed on genomic alterations and gene expression at the mRNA level. However, the molecular basis of CLL variability remains incompletely understood. We speculate that this is because the clinical phenotype is ultimately determined by gene expression at the protein level. We have therefore embarked on a global proteomic study of a large number of CLL trial samples for which whole genome sequencing data will be available through the Genomics England Ltd (GEL) CLL Pilot Project. We anticipate that our study will bridge the gap in our understanding between genotype and clinical phenotype.
Aims
Recently developed mass spectrometric approaches to highly parallel, label-free protein quantification provide a new opportunity to develop diagnostic, prognostic and predictive biomarkers. SWATH (Sequential Windowed Acquisition of all THeoretical fragments) is a data-independent approach which generates a mass spectral library of fragment ions from all detectable peptide precursors. The composite MS/MS spectra are deconvoluted by alignment with a high quality and exhaustive, tissue specific database, whereupon patient samples may be stratified based on the quantitative expression profile of thousands of proteins. Here, we describe generation of a CLL-specific protein database that will provide a permanent reference source for all future trial samples, as indicated in Figure 1.
Methods
Protein extracts were prepared from CLL cells from 14 individual patients in various stages of the disease. Lysates were pooled and digested before being subjected to cation exchange chromatography. Forty fractions were delivered into a TripleTOF 6600 mass spectrometer (SCIEX) via an Eksigent nanoLC 415 system (SCIEX). Data dependent acquisition was performed using 25 MS/MS per cycle to optimise for quality and 30 MS/MS per cycle to optimise for coverage, and the combined data were searched using ProteinPilot 5.0 (SCIEX) against the SwissProt database (Nov 2015, 20,193 human entries). Proteins lying within a 1% global false discovery rate were included in the database.
Results
The new CLL-specific database contains 7773 proteins. The database covers 50% of all human UniProtKB/SwissProt entries which have evidence at the protein level. Pathway analysis of B cell receptor (BCR) signalling based on the GeneGo pathway maps in the MetaCore database (version 6.14, build 61508; Thomson Reuters) showed that this CLL database covers over 87% of the proteins involved in BCR signalling. The quality of the database entries is further underlined by high representation in the Human Genome database (20,814 entries) in aspect of Gene Ontology biological processes and molecular functions, as determined by PANTHER over-representation tests.
Conclusion
Our global protein expression database for CLL cells is thus the most complete database of its type. By combining advanced computational biology to relate protein expression to whole genome sequencing data, the database will provide a framework for elucidating the effect of genomic alterations on protein expression. The construction of this comprehensive CLL database also provides the platform for applying SWATH/MS-based analysis of individual patient samples to identify novel drug targets and predictive biomarkers of disease progression and treatment response.
Session topic: E-poster
Keyword(s): Chronic lymphocytic leukemia, Proteomics
Type: Eposter Presentation
Background
Chronic Lymphocytic Leukaemia (CLL) is notable for its clinical variability, suggesting a role for precision medicine. Attempts at understanding the biological basis of this variability have mainly focussed on genomic alterations and gene expression at the mRNA level. However, the molecular basis of CLL variability remains incompletely understood. We speculate that this is because the clinical phenotype is ultimately determined by gene expression at the protein level. We have therefore embarked on a global proteomic study of a large number of CLL trial samples for which whole genome sequencing data will be available through the Genomics England Ltd (GEL) CLL Pilot Project. We anticipate that our study will bridge the gap in our understanding between genotype and clinical phenotype.
Aims
Recently developed mass spectrometric approaches to highly parallel, label-free protein quantification provide a new opportunity to develop diagnostic, prognostic and predictive biomarkers. SWATH (Sequential Windowed Acquisition of all THeoretical fragments) is a data-independent approach which generates a mass spectral library of fragment ions from all detectable peptide precursors. The composite MS/MS spectra are deconvoluted by alignment with a high quality and exhaustive, tissue specific database, whereupon patient samples may be stratified based on the quantitative expression profile of thousands of proteins. Here, we describe generation of a CLL-specific protein database that will provide a permanent reference source for all future trial samples, as indicated in Figure 1.
Methods
Protein extracts were prepared from CLL cells from 14 individual patients in various stages of the disease. Lysates were pooled and digested before being subjected to cation exchange chromatography. Forty fractions were delivered into a TripleTOF 6600 mass spectrometer (SCIEX) via an Eksigent nanoLC 415 system (SCIEX). Data dependent acquisition was performed using 25 MS/MS per cycle to optimise for quality and 30 MS/MS per cycle to optimise for coverage, and the combined data were searched using ProteinPilot 5.0 (SCIEX) against the SwissProt database (Nov 2015, 20,193 human entries). Proteins lying within a 1% global false discovery rate were included in the database.
Results
The new CLL-specific database contains 7773 proteins. The database covers 50% of all human UniProtKB/SwissProt entries which have evidence at the protein level. Pathway analysis of B cell receptor (BCR) signalling based on the GeneGo pathway maps in the MetaCore database (version 6.14, build 61508; Thomson Reuters) showed that this CLL database covers over 87% of the proteins involved in BCR signalling. The quality of the database entries is further underlined by high representation in the Human Genome database (20,814 entries) in aspect of Gene Ontology biological processes and molecular functions, as determined by PANTHER over-representation tests.
Conclusion
Our global protein expression database for CLL cells is thus the most complete database of its type. By combining advanced computational biology to relate protein expression to whole genome sequencing data, the database will provide a framework for elucidating the effect of genomic alterations on protein expression. The construction of this comprehensive CLL database also provides the platform for applying SWATH/MS-based analysis of individual patient samples to identify novel drug targets and predictive biomarkers of disease progression and treatment response.
Session topic: E-poster
Keyword(s): Chronic lymphocytic leukemia, Proteomics
{{ help_message }}
{{filter}}