
COMBINATION OF RNA- AND EXOME-SEQUENCING EFFICIENTLY ELIMINATES FALSE-POSITIVE SOMATIC POINT MUTATIONS AND INDELS – EXEMPLIFIED BY CASES OF CN-AML
(Abstract release date: 05/19/16)
EHA Library. Hansen M. 06/09/16; 132472; E923

Mr. Marcus Hansen
Contributions
Contributions
Abstract
Abstract: E923
Type: Eposter Presentation
Background
Thorough annotation as a means of detecting highly relevant mutations, and aberrated genes, is becoming more feasible as the evidence of biological pathways underlying malignant transformation compiles. However, there is a continuous risk of misinterpretating both true and false positive observations, which is closely coupled to the sheer number of observations in NGS projects. Thus, there is still a need to find ways to efficiently filter data ab initio, with little knowledge added from disease aetiology and other clinical findings in order to gain new insights.
Aims
We aimed at finding an efficient method to pinpoint true positive and known causal mutations, decreasing the need of downstream interpretation of variants to a minimum.
Methods
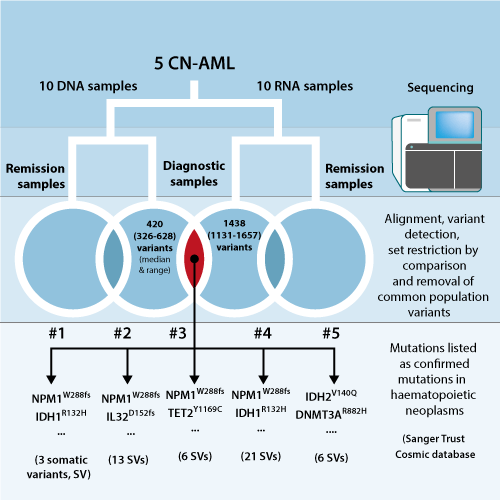
DNA and RNA were extracted from lysated bone marrow aspirate to a mean concentration of 35 ng/µl for both groups and 20 samples in total (Fig.). In order to streamline the alignment and variant calling workflow, we implemented CLC Biomedical Workbench (Qiagen, Aarhus, DK) to keep all dataprocessing in one simple workflow. Only regions covered by the Nextera Rapid Capture Exome kit (Illumina, CA, USA) were included and common variants excluded (dbSNP, NCBI, MD, USA). Alternative GATK and MuTect de facto standard workflow was used to survey the quality of CLC Biomedical Workbench alignment and single nucleotide variant (SNV) detection.
Results
Exome sequencing yielded an average of 9.1*10^7 sequence reads with read lengths of 100 and over 99% mapped in each sample. The combination of whole exome and RNA sequencing efficiently reduced the number of detected mutations to a median number of 6 [3, 21] somatic nucleotide variants (see figure). 6 new mutations, reported to be mutated in at least one other case of haematological neoplasms (COSMIC, Wellcome Trust, Sanger Institute, Cambridge, UK), i.e. not detected by routine laboratory assays at the department. Detection of NPM1 mutations confirmed results from Sanger sequencing, but were found in one additional sample. Likewise, IDH1 census mutation confirmed positive lab results, one additional mutation previously undetected and added IDH2 mutation. CLC Biomedical Workbench did not prove successful in detecting FLT3 internal tandem repeats. The cytogenical status was confirmed by assessment of allelic frequencies from NGS (not shown).
Conclusion
Removal of the substantial number of false positives and irrelevant mutations are a high-priority issues when performing NGS. As CLC Biomedical Workbench, and other tools such as VarScan2, generally detects a large number of somatic variants in raw comparison of paired samples it is crucial to apply stringent and optimal filtering. We show that the inclusion of RNA sequencing in the workflow, not only provides information on malignant expression profiles excluded here, but importantly help to capture the, often very few somatic mutations of myeloid leukaemia. As signature indels, such as found in NPM1, TET2 and FLT3, are important drivers the practicality of DNA-RNA combination is evident, as we demonstrate here – with the exception of FLT3-ITD aberration, which was not detected properly. This small research study not only shows how careful the large number of variants from NGS must be approached – it is also in agreement with previously reported relatively low number of somatic mutations, concluded by several studies. Although we use remission samples in both cases for added stringency, a clinical practical counterpart would be the combination of RNA and DNA malignant sample and skin biopsy or sorted control, DNA only, in whole exome or even targeted panel sequencing.
Session topic: E-poster
Keyword(s): Acute myeloid leukemia, Diagnosis, Somatic mutation
Type: Eposter Presentation
Background
Thorough annotation as a means of detecting highly relevant mutations, and aberrated genes, is becoming more feasible as the evidence of biological pathways underlying malignant transformation compiles. However, there is a continuous risk of misinterpretating both true and false positive observations, which is closely coupled to the sheer number of observations in NGS projects. Thus, there is still a need to find ways to efficiently filter data ab initio, with little knowledge added from disease aetiology and other clinical findings in order to gain new insights.
Aims
We aimed at finding an efficient method to pinpoint true positive and known causal mutations, decreasing the need of downstream interpretation of variants to a minimum.
Methods
DNA and RNA were extracted from lysated bone marrow aspirate to a mean concentration of 35 ng/µl for both groups and 20 samples in total (Fig.). In order to streamline the alignment and variant calling workflow, we implemented CLC Biomedical Workbench (Qiagen, Aarhus, DK) to keep all dataprocessing in one simple workflow. Only regions covered by the Nextera Rapid Capture Exome kit (Illumina, CA, USA) were included and common variants excluded (dbSNP, NCBI, MD, USA). Alternative GATK and MuTect de facto standard workflow was used to survey the quality of CLC Biomedical Workbench alignment and single nucleotide variant (SNV) detection.
Results
Exome sequencing yielded an average of 9.1*10^7 sequence reads with read lengths of 100 and over 99% mapped in each sample. The combination of whole exome and RNA sequencing efficiently reduced the number of detected mutations to a median number of 6 [3, 21] somatic nucleotide variants (see figure). 6 new mutations, reported to be mutated in at least one other case of haematological neoplasms (COSMIC, Wellcome Trust, Sanger Institute, Cambridge, UK), i.e. not detected by routine laboratory assays at the department. Detection of NPM1 mutations confirmed results from Sanger sequencing, but were found in one additional sample. Likewise, IDH1 census mutation confirmed positive lab results, one additional mutation previously undetected and added IDH2 mutation. CLC Biomedical Workbench did not prove successful in detecting FLT3 internal tandem repeats. The cytogenical status was confirmed by assessment of allelic frequencies from NGS (not shown).
Conclusion
Removal of the substantial number of false positives and irrelevant mutations are a high-priority issues when performing NGS. As CLC Biomedical Workbench, and other tools such as VarScan2, generally detects a large number of somatic variants in raw comparison of paired samples it is crucial to apply stringent and optimal filtering. We show that the inclusion of RNA sequencing in the workflow, not only provides information on malignant expression profiles excluded here, but importantly help to capture the, often very few somatic mutations of myeloid leukaemia. As signature indels, such as found in NPM1, TET2 and FLT3, are important drivers the practicality of DNA-RNA combination is evident, as we demonstrate here – with the exception of FLT3-ITD aberration, which was not detected properly. This small research study not only shows how careful the large number of variants from NGS must be approached – it is also in agreement with previously reported relatively low number of somatic mutations, concluded by several studies. Although we use remission samples in both cases for added stringency, a clinical practical counterpart would be the combination of RNA and DNA malignant sample and skin biopsy or sorted control, DNA only, in whole exome or even targeted panel sequencing.
Session topic: E-poster
Keyword(s): Acute myeloid leukemia, Diagnosis, Somatic mutation
Abstract: E923
Type: Eposter Presentation
Background
Thorough annotation as a means of detecting highly relevant mutations, and aberrated genes, is becoming more feasible as the evidence of biological pathways underlying malignant transformation compiles. However, there is a continuous risk of misinterpretating both true and false positive observations, which is closely coupled to the sheer number of observations in NGS projects. Thus, there is still a need to find ways to efficiently filter data ab initio, with little knowledge added from disease aetiology and other clinical findings in order to gain new insights.
Aims
We aimed at finding an efficient method to pinpoint true positive and known causal mutations, decreasing the need of downstream interpretation of variants to a minimum.
Methods
DNA and RNA were extracted from lysated bone marrow aspirate to a mean concentration of 35 ng/µl for both groups and 20 samples in total (Fig.). In order to streamline the alignment and variant calling workflow, we implemented CLC Biomedical Workbench (Qiagen, Aarhus, DK) to keep all dataprocessing in one simple workflow. Only regions covered by the Nextera Rapid Capture Exome kit (Illumina, CA, USA) were included and common variants excluded (dbSNP, NCBI, MD, USA). Alternative GATK and MuTect de facto standard workflow was used to survey the quality of CLC Biomedical Workbench alignment and single nucleotide variant (SNV) detection.
Results
Exome sequencing yielded an average of 9.1*10^7 sequence reads with read lengths of 100 and over 99% mapped in each sample. The combination of whole exome and RNA sequencing efficiently reduced the number of detected mutations to a median number of 6 [3, 21] somatic nucleotide variants (see figure). 6 new mutations, reported to be mutated in at least one other case of haematological neoplasms (COSMIC, Wellcome Trust, Sanger Institute, Cambridge, UK), i.e. not detected by routine laboratory assays at the department. Detection of NPM1 mutations confirmed results from Sanger sequencing, but were found in one additional sample. Likewise, IDH1 census mutation confirmed positive lab results, one additional mutation previously undetected and added IDH2 mutation. CLC Biomedical Workbench did not prove successful in detecting FLT3 internal tandem repeats. The cytogenical status was confirmed by assessment of allelic frequencies from NGS (not shown).
Conclusion
Removal of the substantial number of false positives and irrelevant mutations are a high-priority issues when performing NGS. As CLC Biomedical Workbench, and other tools such as VarScan2, generally detects a large number of somatic variants in raw comparison of paired samples it is crucial to apply stringent and optimal filtering. We show that the inclusion of RNA sequencing in the workflow, not only provides information on malignant expression profiles excluded here, but importantly help to capture the, often very few somatic mutations of myeloid leukaemia. As signature indels, such as found in NPM1, TET2 and FLT3, are important drivers the practicality of DNA-RNA combination is evident, as we demonstrate here – with the exception of FLT3-ITD aberration, which was not detected properly. This small research study not only shows how careful the large number of variants from NGS must be approached – it is also in agreement with previously reported relatively low number of somatic mutations, concluded by several studies. Although we use remission samples in both cases for added stringency, a clinical practical counterpart would be the combination of RNA and DNA malignant sample and skin biopsy or sorted control, DNA only, in whole exome or even targeted panel sequencing.
Session topic: E-poster
Keyword(s): Acute myeloid leukemia, Diagnosis, Somatic mutation
Type: Eposter Presentation
Background
Thorough annotation as a means of detecting highly relevant mutations, and aberrated genes, is becoming more feasible as the evidence of biological pathways underlying malignant transformation compiles. However, there is a continuous risk of misinterpretating both true and false positive observations, which is closely coupled to the sheer number of observations in NGS projects. Thus, there is still a need to find ways to efficiently filter data ab initio, with little knowledge added from disease aetiology and other clinical findings in order to gain new insights.
Aims
We aimed at finding an efficient method to pinpoint true positive and known causal mutations, decreasing the need of downstream interpretation of variants to a minimum.
Methods
DNA and RNA were extracted from lysated bone marrow aspirate to a mean concentration of 35 ng/µl for both groups and 20 samples in total (Fig.). In order to streamline the alignment and variant calling workflow, we implemented CLC Biomedical Workbench (Qiagen, Aarhus, DK) to keep all dataprocessing in one simple workflow. Only regions covered by the Nextera Rapid Capture Exome kit (Illumina, CA, USA) were included and common variants excluded (dbSNP, NCBI, MD, USA). Alternative GATK and MuTect de facto standard workflow was used to survey the quality of CLC Biomedical Workbench alignment and single nucleotide variant (SNV) detection.
Results
Exome sequencing yielded an average of 9.1*10^7 sequence reads with read lengths of 100 and over 99% mapped in each sample. The combination of whole exome and RNA sequencing efficiently reduced the number of detected mutations to a median number of 6 [3, 21] somatic nucleotide variants (see figure). 6 new mutations, reported to be mutated in at least one other case of haematological neoplasms (COSMIC, Wellcome Trust, Sanger Institute, Cambridge, UK), i.e. not detected by routine laboratory assays at the department. Detection of NPM1 mutations confirmed results from Sanger sequencing, but were found in one additional sample. Likewise, IDH1 census mutation confirmed positive lab results, one additional mutation previously undetected and added IDH2 mutation. CLC Biomedical Workbench did not prove successful in detecting FLT3 internal tandem repeats. The cytogenical status was confirmed by assessment of allelic frequencies from NGS (not shown).
Conclusion
Removal of the substantial number of false positives and irrelevant mutations are a high-priority issues when performing NGS. As CLC Biomedical Workbench, and other tools such as VarScan2, generally detects a large number of somatic variants in raw comparison of paired samples it is crucial to apply stringent and optimal filtering. We show that the inclusion of RNA sequencing in the workflow, not only provides information on malignant expression profiles excluded here, but importantly help to capture the, often very few somatic mutations of myeloid leukaemia. As signature indels, such as found in NPM1, TET2 and FLT3, are important drivers the practicality of DNA-RNA combination is evident, as we demonstrate here – with the exception of FLT3-ITD aberration, which was not detected properly. This small research study not only shows how careful the large number of variants from NGS must be approached – it is also in agreement with previously reported relatively low number of somatic mutations, concluded by several studies. Although we use remission samples in both cases for added stringency, a clinical practical counterpart would be the combination of RNA and DNA malignant sample and skin biopsy or sorted control, DNA only, in whole exome or even targeted panel sequencing.
Session topic: E-poster
Keyword(s): Acute myeloid leukemia, Diagnosis, Somatic mutation
{{ help_message }}
{{filter}}