

Contributions
Type: Oral Presentation
Presentation during EHA20: From 12.06.2015 12:15 to 12.06.2015 12:30
Location: Room A8
Background
Genome-wide association studies have identified 37 SNPs in 26 loci that confer risk of CLL. Here, we analyse genomic DNA from a prospective cohort of CLL patients that have an MBL sibling for the presence of these CLL risk alleles.
Aims
To investigate whether CLL risk loci are also prevalent in MBL cells found in a sibling.
Methods
Whole blood of siblings of patients with confirmed CLL patients was screened with informed consent and ethical approval for presence of a distinct CD19+CD5+CD20lowCD79blow population by flow cytometry. If such an MBL population was present, genomic DNA from granulocytes of both CLL patient and MBL sibling was isolated from the same blood sample. All cases with sufficient available DNA were subjected to whole exome sequencing (WES) based on SureSelect Human All Exon V4 kit (Agilent) capture on the HiSeq2000 (Illumina) platform to an average coverage of 40x. Sequence reads were filtered and mapped to the human reference genome (GRCh37). Analysis of gene copy numbers and heterozygosity was performed by either 10K, 250K, SNP6, or CytoscanHD arrays (Affymetrix). In addition, PCR was performed on 35 reported CLL susceptibility alleles and analyzed by direct Sanger sequencing of gel-purified amplicons (PCR Clean-Up System, Promega). The frequency of all risk alleles was compared to healthy individuals from the literature by Fisher’s exact test.
Results
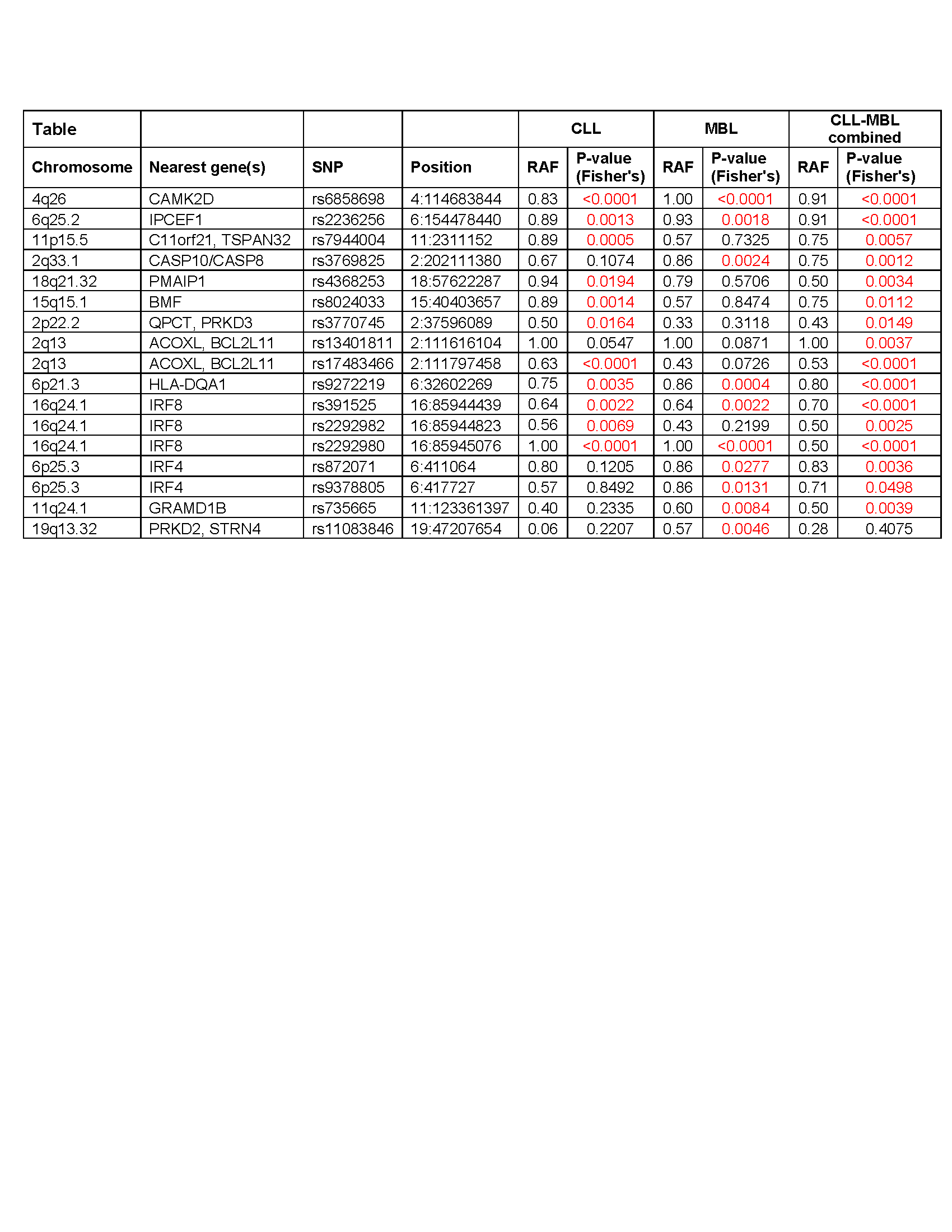
Screening of 160 siblings of CLL patients identified 19 MBL siblings (MBL prevalence: 11.9%) from 16 CLL patients. In all but 2 MBL, the clonal B-cell count was <100/ul. Genomic data from 13 CLL and 14 MBL cases, including 12 complete CLL/MBL pairs are described in this report. Nine of total of 37 CLL susceptibility SNPs could be called on WES data obtained from 5 CLL and 4 MBL cases. 18 CLL risk SNPs could be analyzed on SNP arrays as indicated by a median call rate of at least 98%. The combined SNP array and WES data permitted analysis of 23 SNP of 15 loci. By targeted re-sequencing on 10 CLL and 6 MBL cases, the total number of analyzed risk SNPs was increased to 35 of the 37 known CLL risk alleles. 16 risk alleles were statistically overrepresented in combined CLL and MBL compared to the healthy population (Table). Out of these 16 SNPs, 11 were significantly overrepresented in CLL and nine in MBL, respectively. SNP rs11083846 was significantly overrepresented in MBL only but not in CLL or in the combined CLL and MBL cases.
Summary
Our data provide an independent validation of 16 of 35 previously reported CLL risk loci using combined SNP array, WES, and Sanger sequencing data on a unique prospectively assembled cohort of co-occurrence of CLL and MBL in siblings. No discrepancies were observed between the various analysis platforms. Besides providing genetic correlates for a familial susceptibility in CLL, the shared presence of these risk loci in CLL and their MBL siblings indicate a causal role for clonal premalignant B-cell expansion. These risk alleles therefore appear to contribute to the initiation of a CLL-like phenotype, but further endogenous or exogenous triggers are required to drive MBL to CLL.
Keyword(s): Chronic lymphocytic leukemia, SNP
Session topic: CLL - Biology: Interacting determinants of CLL ontogeny and evolution
Type: Oral Presentation
Presentation during EHA20: From 12.06.2015 12:15 to 12.06.2015 12:30
Location: Room A8
Background
Genome-wide association studies have identified 37 SNPs in 26 loci that confer risk of CLL. Here, we analyse genomic DNA from a prospective cohort of CLL patients that have an MBL sibling for the presence of these CLL risk alleles.
Aims
To investigate whether CLL risk loci are also prevalent in MBL cells found in a sibling.
Methods
Whole blood of siblings of patients with confirmed CLL patients was screened with informed consent and ethical approval for presence of a distinct CD19+CD5+CD20lowCD79blow population by flow cytometry. If such an MBL population was present, genomic DNA from granulocytes of both CLL patient and MBL sibling was isolated from the same blood sample. All cases with sufficient available DNA were subjected to whole exome sequencing (WES) based on SureSelect Human All Exon V4 kit (Agilent) capture on the HiSeq2000 (Illumina) platform to an average coverage of 40x. Sequence reads were filtered and mapped to the human reference genome (GRCh37). Analysis of gene copy numbers and heterozygosity was performed by either 10K, 250K, SNP6, or CytoscanHD arrays (Affymetrix). In addition, PCR was performed on 35 reported CLL susceptibility alleles and analyzed by direct Sanger sequencing of gel-purified amplicons (PCR Clean-Up System, Promega). The frequency of all risk alleles was compared to healthy individuals from the literature by Fisher’s exact test.
Results
Screening of 160 siblings of CLL patients identified 19 MBL siblings (MBL prevalence: 11.9%) from 16 CLL patients. In all but 2 MBL, the clonal B-cell count was <100/ul. Genomic data from 13 CLL and 14 MBL cases, including 12 complete CLL/MBL pairs are described in this report. Nine of total of 37 CLL susceptibility SNPs could be called on WES data obtained from 5 CLL and 4 MBL cases. 18 CLL risk SNPs could be analyzed on SNP arrays as indicated by a median call rate of at least 98%. The combined SNP array and WES data permitted analysis of 23 SNP of 15 loci. By targeted re-sequencing on 10 CLL and 6 MBL cases, the total number of analyzed risk SNPs was increased to 35 of the 37 known CLL risk alleles. 16 risk alleles were statistically overrepresented in combined CLL and MBL compared to the healthy population (Table). Out of these 16 SNPs, 11 were significantly overrepresented in CLL and nine in MBL, respectively. SNP rs11083846 was significantly overrepresented in MBL only but not in CLL or in the combined CLL and MBL cases.
Summary
Our data provide an independent validation of 16 of 35 previously reported CLL risk loci using combined SNP array, WES, and Sanger sequencing data on a unique prospectively assembled cohort of co-occurrence of CLL and MBL in siblings. No discrepancies were observed between the various analysis platforms. Besides providing genetic correlates for a familial susceptibility in CLL, the shared presence of these risk loci in CLL and their MBL siblings indicate a causal role for clonal premalignant B-cell expansion. These risk alleles therefore appear to contribute to the initiation of a CLL-like phenotype, but further endogenous or exogenous triggers are required to drive MBL to CLL.
Keyword(s): Chronic lymphocytic leukemia, SNP
Session topic: CLL - Biology: Interacting determinants of CLL ontogeny and evolution